







Nasjonalt handlingsprogram med retningslinjer for diagnostikk, behandling og oppfølging av kreft hos barn
Forord
Sist faglig oppdatert: 26.05.2020
Mange medisinske faggrupper har i en årrekke lagt ned et betydelig arbeid for å komme frem til konsensusbaserte faglige anbefalinger for diagnostikk og behandling av ulike typer kreft. Som ledd i Nasjonal strategi for kreftområdet (2006–2009) fikk Helsedirektoratet i oppdrag å videreutvikle og oppdatere faggruppenes anbefalinger til nasjonale handlingsprogrammer for kreftbehandling, i nært samarbeid med fagmiljøene, de regionale helseforetakene, Nasjonalt kunnskapssenter for helsetjenesten, og andre relevante myndigheter. De nasjonale handlingsprogrammene representerer en videreføring og en formalisering av faggruppenes anbefalinger.
Nasjonale handlingsprogrammer for kreftbehandling skal bidra til at det offentlige tilbudet i kreftomsorgen blir av god kvalitet og likeverdig over hele landet. Målgrupper for retningslinjene er leger og legespesialister innen medisin, kirurgi, onkologi, radiologi, patologi og fastleger. De vil også være av interesse for andre faggrupper som er involvert i behandling av pasientgruppen og pasienter og pårørende.
Nasjonale retningslinjer fra Helsedirektoratet er å betrakte som anbefalinger og råd, basert på oppdatert faglig kunnskap som er fremskaffet på en systematisk, kunnskapsbasert måte. De nasjonale retningslinjene gir uttrykk for hva som anses som god praksis på utgivelsestidspunktet og er ment som et hjelpemiddel ved de avveininger tjenesteyterne må gjøre for å oppnå forsvarlighet og god kvalitet i tjenesten. Nasjonale retningslinjer er ikke direkte rettslig bindende for mottagerne, men bør langt på vei være styrende for de valg som skal tas. Ved å følge oppdaterte nasjonale retningslinjer vil fagpersonell bidra til å oppfylle kravet om faglig forsvarlighet. Dersom en velger løsninger som i vesentlig grad avviker fra de nasjonale retningslinjene, bør en dokumentere dette og være forberedt på å begrunne sine valg. Sykehusenes eiere og ledelse bør tilrettelegge virksomheten slik at de nasjonale retningslinjene kan følges.
Helsedirektoratet takker arbeidsgruppen for stor innsats i utarbeidelsen av handlingsprogrammet. Vi håper handlingsprogrammet vil være et nyttig arbeidsredskap ved behandling av barn med kreft. Innholdet i handlingsprogrammet vil vurderes årlig og om nødvendig oppdateres.
Disse nasjonale retningslinjene for diagnostikk, behandling og oppfølging av kreft hos barn er publisert 26.05.2020.
Bjørn Guldvog
Helsedirektør
Innledning
Sist faglig oppdatert: 26.05.2020
Av totalt ca 26 000 krefttilfeller i Norge i året utgjør bare ca 140 (0,6 %) barn i alder < 15 år (Nasjonalt kvalitetsregister for barnekreft, 2015). Hvis man tar med aldersgruppen 16–18 år, kommer tallet opp i 180–190. Kreft hos barn skiller seg på flere måter fra kreft hos voksne:
- Kreft hos voksne er ofte definert ut fra organet det går ut fra (lunge, mamma, colon), mens barnekreft defineres etter morfologi og type vev det oppstår i (benmarg, sentralnervesystemet, lymfatisk vev, sympatisk nervesystem, muskel, ben).
- Solide svulster utenfor CNS er oftest embryonale svulster eller sarkomer – hos voksne dominerer carcinomer.
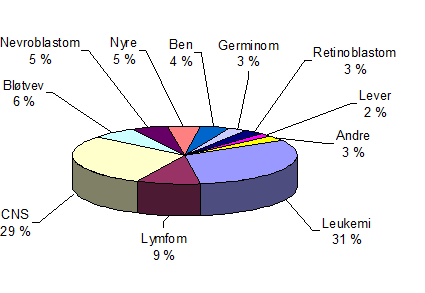
- Krefttypene er forskjellige fra barn til voksne. De vanligste kreftformene hos barn er leukemi og hjernesvulster (figur 1).
- Prognosen ved barnekreft er generelt bedre enn hos voksne, og langtidsoverlevelsen totalt ligger nå på 80 % (Bosetti et al., 2010; Gatta et al., 2014; Kaatsch et al., 2006). Prognosen er avhengig av typen kreft og utbredelse ved diagnosetidspunkt. Ved lymfomer, Wilms tumor og akutt lymfatisk leukemi er overlevelsen godt over 80 %, for retinoblastom > 98 %, mens den ved enkelte solide svulster kan være vesentlig dårligere. Det henvises for øvrig til omtalen av de enkelte sykdomsgruppene
- Behandlingen må ofte gis systemisk, da kreftsykdommen oftest er enten systemisk eller allerede metastasert ved diagnosetidspunktet. Kreft i barnealderen svarer overveiende godt på kjemoterapi, slik at behandlingsresultatene kan være gode selv ved utbredt sykdom. Ved leukemi og lymfomer gis oftest kun kjemoterapi, i enkelte tilfeller supplert med strålebehandling. CNS-svulster blir primært operert dersom dette er mulig. Etterbehandling kan være nødvendig avhengig av svulsttypen og kan bestå av kjemoterapi, strålebehandling eller begge deler, avhengig av pasientens alder. Solide svulster behandles oftest med multimodal terapi, vanligvis med preoperativ kjemoterapi, operasjon og påfølgende kjemoterapi. I utvalgte tilfeller suppleres med strålebehandling og/eller høydosebehandling med stamcellestøtte.
- Ungdom mellom 16 og 18 år begynner å få krefttyper som ligner mer på kreft hos voksne, men særlig ved CNS svulster ser man fortsatt en dominans av de samme krefttypene som hos barn.
Dette handlingsprogrammet er utarbeidet på en litt annen måte enn for andre kreftområder. I stor grad henvises til protokoller – både mht. konkrete behandlingsanbefalinger og litteraturhenvisninger. Dette har man funnet mest hensiktsmessig fordi:
- Behandlingen av barn med kreft i Norge gis i henhold til enten behandlingsprotokoller eller forskningsprotokoller der slike finnes.
- Behandlingsansvaret er regionalisert, og alle som har et behandlingsansvar kjenner til de aktuelle protokollene.
- Protokollene er utarbeidet på bakgrunn av en kunnskapsbasert prosess.
- Protokollene inneholder alle sentrale litteraturhenvisninger.
- At behandling gis etter slike protokoller, sikrer at alle barn i landet får lik behandling etter internasjonalt aksepterte normer, at man kan høste felles erfaring og øke kunnskapen om de forskjellige tilstandene.
- Kreft hos barn omfatter en lang rekke sjeldne tilstander. Per i dag brukes flere enn 30 protokoller. For noen tilstander diagnostiseres færre enn 1 barn pr år i Norge, og det er derfor ikke hensiktsmessig å omtale alle aktuelle tilstander i detalj.
- Fordi mange tilstander er svært sjeldne og det er stor heterogenitet blant pasientene, er evidensbasen langt mindre enn hva tilfellet oftest er i voksenonkologien.
- Av den grunn kan man ikke gjøre kost-nyttevurderinger på samme måte som i voksenonkologien.
Norsk barnekreftbehandling er regionalisert ved at hvert regionsykehus har ansvar for kreftbehandlingen i sin region. Allogen stamcelletransplantasjon, organtransplantasjon og retinoblastombehandling er sentralisert til OUS. Behandling med strålekniven (gammakniv) er sentralisert til Haukeland Sykehus. Utredning, operasjoner, strålebehandling og mesteparten av kjemoterapien skjer ved regionsavdelingene, mens mye av støttebehandlingen (infeksjonsbehandling, transfusjoner) samt deler av cellegiftbehandlingen foregår ved lokale barneavdelinger.
Barnekreftbehandling er uttalt tverrfaglig, og ved hvert regionsykehus er det tverrfaglige team med bred sammensetning for solide svulster i og utenfor sentralnervesystemet. Myndighetene etablerte Kompetansesenter for solide svulster hos barn (KSSB – fra 2012 «Nasjonal kompetansetjeneste for solide svulster hos barn») i 1999 ved Rikshospitalet, for å koordinere et nasjonalt faglig nettverk innen barnekreft. Innen Kompetansetjenesten er det etablert to faggrupper for svulster henholdsvis i og utenfor sentralnervesystemet. Begge faggruppene er bredt faglig og regionalt sammensatt. Faggruppene utpeker nasjonale koordinatorer for de enkelte svulstene og deltar i internasjonalt protokollsamarbeid. Faggruppene bestemmer hvilke protokoller man skal tilsluttes nasjonalt. De fleste protokoller er nå utarbeidet av den europeiske gren av den Internasjonale barnekreftorganisasjon SIOP (SIOP-E). Kompetansetjenesten har gjennom faggruppene ansvaret for at alle pasientene får likeverdig behandling gjennom nasjonalt samarbeid, uansett hvor de bor.
Leukemibehandlingen er organisert gjennom Norsk barneleukemigruppe (NBLG) og Nordisk forening for pediatrisk hematologi og onkologi (NOPHO), som har utarbeidet felles nordiske protokoller som Norge er tilsluttet.
Siden mange av barnekreftformene er så sjeldne at det kun diagnostiseres færre enn 5 nye pasienter årlig, er det ikke overkommelig å søke formell godkjenning for hver enkelt protokoll som benyttes. Av den grunn vil det i en del tilfeller benyttes internasjonale behandlingsprotokoller som «best available treatment». Hvis det er randomiseringer i slike protokoller brukes standardarmen.
Ungdom opp til 18 år med kreft skal nå behandles på barne- og ungdomsavdelinger dersom det ikke er spesielle grunner for noe annet (f. eks. eldre tenåringer med typisk «voksne» kreftformer slik som testikkelkreft). Disse avdelingene må ha tilrettelagt forholdene etter ungdommenes spesielle behov.
Alle barn med kreft i Norge får nå tilbud om den samme behandlingen og blir dersom det er aktuelt, tilbudt å delta i internasjonale forskningsprotokoller. Hvert senter utpeker ansvarlige for organiseringen lokalt.
All barnekreft meldes til Kreftregisteret via det elektroniske meldesystemet KREMT. Klassifiseringen av barnekreft har fulgt egne retningslinjer siden 1987 og er basert på ICD-O. Diagnostiske grupper er basert mest på histologi/morfologi, og det er definert 12 hovedgrupper. Lokalisasjon blir registrert. Siden 2001 er det definert et eget barnekreftregister («Norsk kvalitetsregister for barnekreft», som ligger under kreftregisteret) som også registrerer behandling og oppfølging. Pasientene som behandles etter internasjonale protokoller må i tillegg registreres i studiedatabasene. For leukemiene skjer dette online i sammenheng med de felles nordiske protokollene.
Flere internettsteder inneholder retningslinjer for utredning og behandling av kreft hos barn. Ofte inneholder disse steder en offentlig og en passordbelagt lukket del forbeholdt medlemmer/barneonkologer. Behandlingsprotokollene ligger vanligvis i den lukkede delen. Det kan søkes om tilgang til den lukkede delen. De aktuelle protokollene er tilgjengelige for de leger som behandler pasienten.
Aktuelle nettsteder er:
- Kompetansesenter for solide svulster hos barn (KSSB) www.kssb-no.org
- Nordisk forening for pediatrisk hematologi og onkologi (NOPHO): https://www.nopho.net/
- SIOP (International Society of Paediatric Oncology): www.siop.nl
Epidemiologi
Sist faglig oppdatert: 26.05.2020
Insidensen av nye tilfeller i Norge ligger med 15 nye tilfeller per 100 000 barn/år (alder < 15 år) omtrent på samme nivå som i resten av Europa (Nasjonalt kvalitetsregister for barnekreft, 2015). Insidensen har vært stabil i Norden de siste 20 år, mens internasjonale undersøkelser synes å tyde på lett stigende tendens (Kaatsch et al., 2006). Leukemier og svulster i sentralnervesystemet utgjør ca 30 % hver (ca 40–50 nye tilfeller hvert år), de resterende svulstene utgjør enkeltvis fra 1–7 %, totalt ca 60 nye tilfeller i året (figur 1). Kreft hos barn er hyppigst i aldersgruppen 0 til 6 år. Nevroblastom og nyresvulster oppstår særlig de første to leveår, leukemi er hyppigst i alderen to til fem år, mens bensvulster og Hodgkins lymfom er vanligst i tenårene. CNS-svulster og bløtvevstumores er mer jevnt fordelt utover hele barnealderen (Kaatsch et al., 2006).
Prognosen ved barnekreft har økt fra omkring 10 % overlevelse før 1970 til nærmere 80 % de seneste år, og de Nordiske land ligger i toppsjiktet internasjonalt mtp behandlingsresultater (Bosetti et al., 2010; Gatta et al., 2005; Gatta et al., 2009).
Lite er kjent om årsak til barnekreft. Så langt synes miljøfaktorer, elektromagnetisk stråling, sosioøkonomisk status av foreldre eller infeksjoner å bety svært lite. Terapeutisk stråling øker faren for sekundær kreft i det bestrålte området på lang sikt. Enkelte cytostatika øker også faren for sekundær kreft. Visse medfødte tilstander øker risikoen for å utvikle kreft i barneårene. Mest kjent er dette for Downs syndrom (akutte lekemier). Andre slike tilstander er ataxia teleangiectatica, Nevrofibromatose type 1 og 2, Fanconi syndrom og en rekke svært sjeldne andre tilstander.
Forebygging
Sist faglig oppdatert: 26.05.2020
Siden årsak til kreft hos barn ikke er klarlagt, er det få muligheter til å forebygge kreft hos barn. Et viktig tiltak er begrensning av diagnostisk (CT) og terapeutisk stråling hos barn.
Forløpstider
Sist faglig oppdatert: 26.05.2020
1. september 2015 ble Pakkeforløp for kreft hos barn innført.
Om pakkeforløp for kreft
Sist faglig oppdatert: 26.05.2020
Pakkeforløp for kreft skal gi forutsigbarhet og trygghet for pasient og pårørende, og er et standard pasientforløp som beskriver organisering av utredning og behandling, kommunikasjon/dialog med pasient og pårørende samt ansvarsplassering og konkrete forløpstider. Pakkeforløpet starter når et helseforetak eller privat ideelt sykehus mottar en henvisning med begrunnet mistanke om kreft, eller når helseforetaket selv starter utredning med begrunnet mistanke om kreft.
Formålet med Pakkeforløp for kreft er at kreftpasienter skal oppleve et godt organisert, helhetlig og forutsigbart forløp uten unødvendig ikke-medisinsk begrunnet forsinkelse i utredning, diagnostikk, behandling og rehabilitering.
Forløpstidene i pakkeforløpet beskriver den maksimale tiden de ulike fasene i forløpet bør ta. Forløpstidene angis i kalenderdager. De enkelte fasenes forløpstid legges til slutt sammen til en samlet forløpstid, som angir tiden fra henvisning er mottatt til behandling er startet. Med utgangspunkt i pakkeforløpet skal et individuelt forløp tilrettelegges for hver enkelt pasient.
De regionale helseforetakene har det overordnede ansvaret for å sikre at pakkeforløpene med forløpstidene blir implementert og fulgt opp. Forløpstidene er normerende og er ikke en pasientrettighet. Fortsatt er det lovmessige grunnlaget pasientrettighetsloven § 2-2 og forskrift om prioritering av helsetjeneste. Av og til vil det av faglige grunner være noen pasienter som ikke kan utredes ferdig innen normert forløpstid for oppstart av første behandling. Årsaker til avvik fra de normerte forløpstidene bør dokumenters i pasientjournalen.
Forløpstider for kreft hos barn
Sist faglig oppdatert: 26.05.2020
| Fra henvisning mottatt til første fremmøte utredende avdeling |
| 3 kalenderdager * |
| Fra første fremmøte i utredende avdeling til avsluttet utredning (beslutning tas) |
| 10 kalenderdager ** |
| Fra avsluttet utredning til start behandling | Kirurgisk behandling | 14 kalenderdager |
| Fra avsluttet utredning til start behandling | Medikamentell behandling | 3 kalenderdager |
| Fra avsluttet utredning til start behandling | Strålebehandling | 14 kalenderdager |
| Fra henvisning mottatt til start behandling | Kirurgisk behandling | 27 kalenderdager |
| Fra henvisning mottatt til start behandling | Medikamentell behandling | 16 kalenderdager |
| Fra henvisning mottatt til start behandling | Strålebehandling | 27 kalenderdager |
*Ved mistanke om leukemi bør pasienten mottas innen 1 kalenderdag
**Ved mistanke om leukemi bør denne forløpstid være maksimalt 4 kalenderdager
**Ved mistanke om lymfekreft på halsen bør denne forløpstid være maksimalt 7 kalenderdager
Pakkeforløp for kreft hos barn finnes på Helsedirektoratets nettsider.
Det er utarbeidet egne diagnoseveiledere for fastleger for inngang til pakkeforløp. Diagnoseveileder for kreft hos barn finnes på Helsedirektoratets nettsider.
Tidlig diagnostikk/screening
Sist faglig oppdatert: 26.05.2020
Til nå er det ikke etablert gode tiltak for tidlig diagnostikk og screening ved barnekreft. Forsøk med screening av nevroblastom har ikke vært vellykket og er nå ikke i bruk internasjonalt.
Diagnostisering
Symptomer, utredning, stadieinndeling
Sist faglig oppdatert: 26.05.2020
Symptomer
Symptomene ved kreft hos barn kan variere sterkt, ut fra svulsttypen, og de fleste symptomene er uspesifikke.
- Leukemi og lymfom: Ved leukemi er de mest framtredende symptomene blekhet og slapphet, hudblødninger, lymfeknutehevelser, feber og smerter i hele kroppen, ofte lokalisert til ledd og skjelett. Dyspnoe, noen ganger livstruende, kan oppstå på grunn av kompresjon av luftveiene ved en mediastinaltumor. Lymfomer starter oftest med en palpabel og ofte synlig lokalisert lymfeknutehevelse som ikke går tilbake. Lymfomer kan også føre til nattesvette og vekttap.
- Hjernesvulster kan medføre økt hjernetrykk, med kvalme, oppkast og hodepine, ofte mest uttalt om morgenen. Hos spedbarn kan unormal økning av hodeomkretsen være første tegn på hjernesvulst. Andre symptomer, forårsaket av tumors lokalisasjon, er krampeanfall, ataksi, fokale nevrologiske utfall og endokrine symptomer som vekstforstyrrelser eller diabetes insipidus.
- Solide svulster fremkaller symptomer ut fra tumors beliggenhet og størrelse. Ikke sjelden er første symptom en synlig eller palpabel tumor, enten i abdomen eller andre steder i kroppen. Skjelettsmerter og -forandringer som noen ganger mistolkes som osteomyelitt, kan forekomme ved beinsarkomer og ved skjelettmetastaser, for eksempel ved nevroblastom. Andre symptomer kan være urinretensjon, hematuri, obstipasjon, nevrologiske symptomer pga kompresjon av medulla eller perifere nerver, hypertensjon.
Utredning
Utredningen ved mistanke om barnekreft foregår etter lignende retningslinjer som hos voksne, men mange av undersøkelsene må foretas i narkose (benmargsundersøkelse, spinalpunksjon, MR, cytologi/biopsi). Når diagnosen er etablert, suppleres med undersøkelser som er spesifisert i de enkelte behandlingsprotokoller (ut fra metastaseringsmønster og planlagt terapi).
I tillegg til anamnese og generell undersøkelse inkl. blodtrykk kan utredningen omfatte:
- Leukemi/lymfom: Benmargsundersøkelse (utstryk/morfologi, immunfenotyping, cytogenetikk, molekylærgenetiske undersøkelser, biopsi), røntgen thorax, ultralyd abdomen.
- Solide svulster: Billeddiagnostikk (MR, CT, ultralyd, rtg thorax, ev. skjelettrøntgen, skjelettscintigrafi, PET scan, MIBG scintigrafi). Cytologi/biopsi (alltid; oftest ønskes både cytologi og histologi). Blodprøver inkl. tumormarkører (HCG, AFP, NSE, ferritin, LD). Urin katekolaminer (HVA, VMA) ved mistanke om nevroblastom. For å kartlegge metastasering gjøres ved noen svulster benmargsundersøkelse.
CNS-svulster: MR undersøkelse er hovedregel og i de fleste tilfeller skal det også utføres MR undersøkelse også av spinalkanalen. Ved mistanke om germinom tas HCG og AFP i blod og spinalvæske. Det nevrokirurgiske inngrepet er ofte primærbehandlingen ved hjernesvulst hos barn. Hos vel halvparten av barn med svulstr i hjerne eller ryggmarg vil det være endelig behandeling.
Med tanke på behandlingens bivirkninger må det gjøres en bred generell utredning før behandlingsstart, avhengig av behandlingens karakter.
For utdypende generell informasjon om symptomer og utredning ved barnekreft henvises til oversiktsverk om pediatrisk hematologi/onkologi (Cancer in children: clinical management, 2005; Principles and practice of pediatric oncology, 2006).
Radiologisk utredning
Ved utredning av solide svulster hos barn er radiologisk diagnostikk av god kvalitet en forutsetning for videre utredning og behandling. Primærutredning lokalt bør begrenses til nødvendig diagnostikk for å gi begrunnet mistanke om malignitet. For svulster i abdomen er ultralyd tilstrekkelig som primærutredning før pasienten sendes til barneonkologisk enhet, hvor resten av den radiologiske utredningen bør utføres. Ved mistanke om tumor i skjelett og thorax er røntgen førstevalg. Det er en potensiell fare for å påføre pasientene sekundære kreftformer som følge av ioniserende stråling, særlig ved computer tomografi (CT)-undersøkelser og etter strålebehandling. Derfor bør undersøkelser tilpasset tumorutredning på barn utføres ved den barneonkologiske enheten. Da små barn oftest må utredes under generell anestesi, er det viktig å planlegge hvilke undersøkelser som er hensiktsmessig å gjøre i samme narkose. Dette gjøres i samarbeid mellom onkolog, radiolog, anestesiolog og kirurg.
Faste opplegg i den radiologiske utredningen må følges, og disse prosedyrer styres av ulike behandlingsprotokoller (nasjonale og internasjonale) for forskjellige tumorformer. Selv om man ikke har endelig histologiske diagnose vil tumors lokalisasjon og utseende ofte styre den primære utredningen.
Generelle anbefalinger
Ultralyd
Ved alle solide tumores i absomen, hals og bløtdeler er ultralyd førstehånds valg for å stadfeste tumors lokalisasjon og forhold til andre organer (kontakt, innvekst, dislokasjon, kompresjon). Farge- og evt spektral doppler er en viktig del av diagnostikken, og utføres for å vurdere vaskularisering av tumor, innvekst eller kompresjon av store kar og evt tumortrombe. Tumormål i tre plan med volumbergening for senere å vurdere terapirespons må alltid utføres. Da barnetumores ofte er svært store når de diagnostiseres, er ultralyd sjelden tilstrekkelig for å vurdere størrelse og forholdet til naboorganer.
Magnetisk resonons tomogtafi (MR)
Alle sykehus i Norge som behandler barn med kreft har oppdaterte MR maskiner. Etter ultralyd eller røntgen er alltid utredning med MR første valg. Generell anestesi eller sedasjon er ofte nødvendig på mindre barn (<6 år). Protokoller for å undersøke tumors lokalisasjon og eventuell lokal spredning eller metastasering er en del av primærutredningen. Primærtumor måles i tre plan med volumberegning for å senere kunne vurdere terapirespons.
Computer tomografi (CT)
Ved de fleste tumores er CT volumopptak av thorax/lunger en obligatorisk del av metastaseutredningen, da MR foreløpig ikke er like god som CT for å detektere små lungemetastaser. I mange internasjonale behandlingsprotokoller står det at røntgen av thorax er tilstrekkelig, men i praksis gjøres det i Norge CT av thorax i tillegg til røntgen.
Positron emisjonstomografi med CT (PET-CT): Har sin plass i utredningen av visse typer tumores (se kapittel for de enkelte diagnoser).
Røntgen
Røntgen av thorax utføres alltid i primærutredningen. Ved bentumores skal det alltid tas vanlig røntgen av affiserte ben i tillegg til MR og evt. CT.
Behandlingskontroller og oppfølging
Alle behandlingsprotokoller har et fast intervall for radiologisk oppfølging. Med den raske teknologiske utviklingen ser man at relativt nye undersøkelsesmodaliteter (PET-CT/MR) legges til protokoller uten at man i tilstrekkelig grad har vurdert/evaluert om andre undersøkelser da kan/bør utgå. Det må derfor være en kontinuerlig dialog mellom radiolog og kliniker om hvilke undersøkelsesmodaliteter som skal benyttes for å redusere strålebelastningen til barnet og for å unngå evt. unødige undersøkelser.
Anbefalinger:
- Radiologisk utredning av solide tumores med MR/CT/PET-CT bør utføres på barneonkologisk enhet ut fra de forskjellige fastsatte protokoller.
- Da små barn oftest må utredes under generell anestesi, må det planlegges nøye slik at eventuell radiologisk og histopatologisk utredning kan utføres i samme narkose.
- Radiologiske kontroller utføres etter behandlingsprotokoller, og i nært samarbeid mellom kliniker og radiolog for å redusere strålebelastningen og for å unngå unødige undersøkelser.
Stadieinndeling
Stadieinndeling eller risikogruppe har betydning for mange kreftformer. Det henvises til de enkelte behandlingsprotokollene.
Patologisk-anatomisk diagnostikk av barnekreft
Sist faglig oppdatert: 26.05.2020
Kreft hos barn kan inndeles i 2 hovedgrupper: leukemier og solide svulster. Den patologisk-anatomiske utredning er prinsipielt basert på 3 områder: morfologi, immunfenotype og genetikk som undersøkes suksessivt etter hverandre avhengig av de diagnostiske og kliniske behov i et gitt tilfelle. Dette gjelder både for leukemier og solide svulster, men metodene kan variere med type prøver som er tatt.
Leukemier
Morfologi: Utstryk fra benmarg og blod vurderes cytologisk. Det tas i tillegg benmargsbiopsi for histologisk undersøkelse. Biopsien er viktig for å vurdere cellulariteten og infiltrasjonsgraden. Konklusjonen fra de morfologiske undersøkelser er som regel at det enten er overbevisende forandringer som ved leukemi, at resultatet er usikkert eller klart negativt. Ved positivt eller usikkert resultat er det helt nødvendig å gå videre med immunfenotyping.
Spinalvæske undersøkes på forekomst av celler (telling).
Immunfenotype: Celler fra benmargsaspirat og/eller blod undersøkes med flowcytometri på normal eller unormal ekspresjon av antigener både på overflaten og inne i cellene. Med denne teknikken kan man analysere mange tusen celler på kort tid. Metoden gir svært begrenset morfologisk informasjon, men det er mulig å se på forekomsten av flere antigener i samme celle. Dette kan være avgjørende for å komme frem til en korrekt diagnose og for å etablere et fenotypisk «vindu» som er karakteristisk for en individuell pasient, og som kan brukes for senere analyser i oppfølgingen av pasienten etter kjemoterapi.
Immunhistologiske undersøkelser av benmargsbiopsi er ikke nødvendige hvis leukemien er tilfredsstillende klassifisert ved hjelp av flowcytometri.
Celler i spinalvæsken immunfenotypes enten immuncytologisk på cytospinpreparater eller med flowcytometri. Etter internasjonal overenskomst trenger man minst 15 positive «events» (dvs. celler) for å kalle et flowcytometrisk resultat for positivt. Dette kan by på problemer når celletallet i spinalvæsken er svært lavt. Her vil en immuncytologisk undersøkelse kunne gi et resultat som er basert på en kombinasjon av cytomorfologi og immunfenotype.
Genetikk: Leukemicellene kan undersøkes ved hjelp av klassisk cytogenetikk, in situ hybridisering eller molekylærgenetiske metoder som ofte er basert på PCR-teknikker. Hovedfordelen med klassisk cytogenetikk er at den representerer en pan-genomisk metode som analyserer alle kromosomer for amplifikasjoner, tap eller translokasjoner av genmateriale. Ulempen er at metoden er avhengig av å dyrke cellene for å høste metafaser, noe som tar for lang tid (uker) i forhold til start av behandling. Noen genetiske avvik som er viktige i vurderingen av leukemier, er dessuten submikroskopiske slik at de ikke sees ved analysen av kromosombåndene.
In situ hybridisering og ulike PCR-metoder er skreddersydd for å analysere kjente genforandringer med en spesifisitet og sensitivitet som er høyere enn klassisk cytogenetikk. Analysen er rask (1–2 dager) og lett å standardisere, men har i forhold til cytogenetikken den ulempen at man kun får svar på det man har spurt om, og ikke registrerer andre eventuelle genetiske abnormiteter.
Både lymfatiske og myelogene leukemier er kjent for en rekke genetiske forandringer som er assosiert med prognosen, og som derfor brukes til valg av behandling. Noen behandlingsformer er rettet spesifikt mot proteiner som er til stede eller forandret på grunn av en genetisk aberrasjon. Derfor utføres i dag en rekke genetiske standardundersøkelser enten ved hjelp av PCR eller in situ-hybridisering når leukemien er flowcytometrisk klassifisert.
Undersøkeler på MRD (minimal residual disease) etter behandling utføres flowcytometrisk og molekylærgenetisk (PCR) avhengig av den immunologiske og genetiske fenotypen som ble etablert på diagnosetidspunktet.
Anbefalinger:
- For leukemier må utstryk fra både benmarg og blod vurderes morfologisk.
- Celler fra benmargsaspirat og/eller blod undersøkes med flowcytometri med henblikk på normal og unormal ekspresjon av antigener både på overflaten og intracellulært.
- Immunhistologiske undersøkelser av benmargsbiopsi er nødvendig der leukemien ikke er tilfredsstillende klassifisert ved hjelp av flowcytometri.
- Leukemicellene undersøkes ved klassisk cytogenetikk, in situ hybridisering og/eller ulike PCR‑metoder for kjente genforandringer.
- Kravene til disse undersøkelser er nedfelt i internasjonale behandlingsprotokoller som oppgraderes fortløpende.
Solide tumores
Morfologi: Biopsier fra solide svulster vurderes primært histologisk i motsetning til prøver fra leukemier der den cytologiske analysen er sentral. Biopsier kan tas med grovnål (tru-cut) eller som skjærebiopsi. Grovnålsbiopsier medfører ingen post-operative komplikasjoner som man kan se etter skjærebiopsier (f. eks. hematogen utsæd av tumorceller eller adheranser ved abdominale svulster), men har klare ulemper: Materialet er ofte svært sparsomt og kan ikke vurderes for representativitet før etter 1–2 dager på grunn av innstøping og skjæring av histologiske snitt.
Derfor anbefales en kombinert cytologisk (finnålsaspirasjon, FNAC) og histologisk (grovnål) undersøkelse. Finnålsaspirasjonen utføres først og som regel under ultralydveiledning. Utstryk fra disse celler farges umiddelbart (3–5 minutter) og vurderes av cytolog mens pasienten venter i narkose. Fargingen gir opplysninger om prøvens representativitet slik at det samme området av lesjonen kan brukes til grovnålsbiopsi. Det er viktig å utføre minst 4 cytologiske aspirasjoner og 4 grovnålsbiopsier for å sikre tilstrekkelig materiale til spesialundersøkelser som vil variere med den kliniske problemstillingen og resultatet fra den første cytologiske hurtigfarging. To av biopsiene støpes i parafin, mens de andre dypfryses for fenotypiske og genetiske spesialundersøkelser som ikke kan utføres på parafinsnitt. Av det cytologiske finnålsaspiratet lages cytospinpreparater som er velegnet til immunfenotypiske og interfase-cytogenetiske undersøkelser. Aspirerte celler kan også dypfryses til senere ekstraksjon av RNA, DNA og proteiner.
Immunfenotype: Ved mange solide svulster kjenner man til en karakteristisk kombinasjon av cellulære antigener som kan analyseres ved hjelp av immuncytologi (f. eks. cytospin fra FNAC) eller immunhistologi av biopsimateriale. Slike undersøkelser er standardprosedyrer i alle patologisk-anatomiske laboratorier.
Genetikk: Genetiske undersøkelser ved solide svulster har flere formål: De kan brukes diagnostisk for å påvise genetiske aberrasjoner, f. eks. translokasjoner som er karakteristiske for en bestemt entitet. Diagnostiske tester er spesielt viktige ved enkelte bløtdelssvulster der man ikke kjenner til en spesifikk immunfenotype I tillegg finnes en rekke genetiske forandringer, f. eks. mutasjoner eller kopitallsforandringer (amplifikasjoner/delesjoner) som har prognostisk og/eller terapeutisk betydning, og som derfor er med å bestemme behandlingen. Helgenomisk sekvensering av solide svulster har avslørt ulike typer mutasjoner som f. eks. av ALK genet i neuroblastom eller B-RAF genet ved Langerhanscelle histiocytose som kan utnyttes til personrettet behandling.
Translokaksjoner undersøkes med in situ hybridisering (FISH) eller PCR-basert påvisning av et fusjonsgen. Kopitallsvorandringer påvises med in situ hybridisering av enkeltgener eller helgenomisk med arrayCGH. Mutasjoner undersøkes med Sanger sekvensering eller – hvis høy sensitivitet er påkrevet – med massiv parallel (dyp) sekvensering (NGS). Det finnes kommersielle platformer som muliggjør en synkron analyse av fusjoner, mutasjoner og kopitallsforandringer av flere titalls gener som er relevante i diagnostikken av solide barnesvulster.
Klassisk cytogenetikk har i de senere år fått mindre betydning i utredningen av solide svulster av årsaker som er belyst ovenfor. Dyrkning av celler fra solide svulster til metafase er ofte vanskeligere ved solide svulster enn ved celler fra blod og benmarg.
In situ-hybridisering og PCR-baserte metoder gir raske og pålitelige svar. Både in situ-hybridisering, arrayCGH og mange PCR-baserte analyser av enkeltgener inkludert NGS kan utføres på formalin-fiksert parafin-innstøpt (FFPE) materiale. Dette innebærer at andelen tumorceller i en prøve kan bestemmes på histologiske snitt før de molekylærgenetiske analyser i gangsettes. Tilblanding av normale celler kan være problematisk når man velger ekstraksjons-baserte metoder. Et ukjent antall ikke-tumorceller i en prøve kan gi falsk negative resultater. Det er derfor påkrevet at en patolog har analysert prosentandelen tumorceller i et cytologisk eller histologisk materiale før dette leveres til molekylærgenetikeren for ekstraksjon av f. eks. RNA eller DNA.
Anbefalinger:
- Biopsier fra solide svulster vurderes primært histologisk. Det anbefales en kombinert cytologisk (finnålsaspirasjon, FNAC) og histologisk (grovnål) undersøkelse. Biopsi tas som regel ultralydveiledet.
- Karakteristiske kombinasjoner av cellulære antigener for solide svulster analyseres ved hjelp av immunhistologi og immuncytologi.
- Både gensekvensering, in situ-hybridisering og arrayCGH utføres avhengig av type tumor for å konfirmere den histologiske diagnosen og/eller vurdere prognosen/velge adekvat behandling.
- Analysering av prosentandel tumorceller i et cytologisk eller histologisk materiale må gjøres før videre molekylærgenetisk testing.
Kravene til disse undersøkelser er nedfelt i tumor-spesifikke, internasjonale behandlingsprotokoller som oppgraderes fortløpende.
Integrert diagnostikk
Sist faglig oppdatert: 26.05.2020
Morfologiske, immuncyto- og histologiske og in situ-hybridiseringer utføres og analyseres på patologisk-anatomiske laboratorier. Flowcytometriske og molekylærgentiske metoder er derimot ofte allokert til spesiallaboratorier. Deres resultater er imidlertid del av en integrert diagnostikk som patologen bør ha hovedansvaret for. De viktigste argumenter for dette er:
- Før materialet sendes til molekylærgenetisk undersøkelse, må patologen garantere representativiteten av materialet og i noen tilfeller kvantifisere prosentandelen tumorceller.
- Resultatene fra alle spesialundersøkelsene bør integreres i den morfologiske besvarelsen for å unngå misforståelser hos rekvirenten (klinikker).
Biobank
Sist faglig oppdatert: 26.05.2020
Ved utredning, diagnostikk og behandling av kreft hos barn er prøvetagning til diagnostiske biobanker sentralt, i form av blod- og benmargsutstryk, benmargsaspirat, cerebrospinalvæske og biopsier fra tumorvev. Det er vesentlig at de vevsprøvene som lagres i patologiavdelingenes diagnostiske biobanker, ivaretas på en slik måte at overskuddsmateriale kan brukes til supplerende undersøkelser dersom ny kunnskap viser at dette kan få konsekvenser for utredning, diagnostikk eller behandling, og til forskningsdrevet behandlingsutvikling.
I tillegg til de diagnostiske biobankene har man i dag en forskningsbiobank for barnekreft; Den Nasjonale Barnekreftbiobanken (NBB). NBB er en samling av biologisk vev (tumorvev, blodprøver, spinalprøver, benmargsprøver, hår, urin og avføring) og data fra barn diagnostisert med kreft. Biobanken ble etablert ved Oslo Universitetssykehus i 2017 (OUS – Barnebiobank-Barnekreft), deretter utvidet til nasjonal biobank (den Norske Barnekreftbiobanken NBB) i 2019. NBB er godkjent som en generell forskningsbiobank av Regional komite for medisinsk og helsefaglig forskningsetikk (REK), ref 2016/943/REK sør-øst. Oslo Universitetssykehus er forskningsansvarlig institusjon for biobanken. Biobanken er forankret i et nasjonalt konsortium (Nasjonalt konsortium for barnekreftbiobanken) og er basert på en konsortieavtale mellom partene som representerer våre 4 helseregioner. Hovedformålet med innsamling av materiale i NBB er forskning som kan bidra til økt forståelse av etiologi, patogenese, forebygging, diagnostikk, behandling, omsorg eller oppfølgning av barnekreft. Utlevering/bruk av biologisk materiale/data til forskningsprosjekter skal ligge innenfor formålet med biobanken slik det er beskrevet, og skal være i tråd med forskningsprotokollen og nødvendige godkjenninger som gjelder for de enkelte forskningsprosjekter som søker om å få utlevert/bruke biologisk materiale/data fra NBB. Informasjon om NBB og søknadsprosessen for utlevering av materiale ligger på nettsiden.
Genetikk
Arv og kreft hos barn
Sist faglig oppdatert: 26.05.2020
Bakgrunn
Kreftceller har genetiske forandringer (mutasjoner), men ved kreft i barnealder er disse vanligvis ervervet. Dette betyr at de ikke er til stede i kimbanen. Kreft i barnealder er derfor vanligvis ikke arvelig. Mange av de ervervete genforandringene i svulster gir opphav til medfødte syndromer når de samme genene er mutert i kimbanen. Det er på denne måten biologiske sammenhenger både mellom regulering av cellevekst og celledifferensiering i fosterlivet, kreftutvikling og medfødte utviklingsavvik. Disse sammenhengene ses igjen klinisk idet man ved mange medfødte syndromer ser kreftdisposisjon (i større eller mindre grad).
- Det er av interesse å identifisere det mindretallet av barnekreftpasienter som har genetisk (hoved‑)årsak til sin svulstsykdom fordi:
- Det har en egenverdi å vite årsaken til at barnet fikk kreftsykdommen
- I noen tilfeller har genetisk betinget kreft en annen prognose enn annen kreft og/eller skal ha annen behandling (eksempel: Arvelig retinoblastom skal i utgangspunktet ikke gis ekstern strålebehandling)
- I noen, men ikke alle, tilfeller, har personer med genetisk kreft høy risiko for å utvikle ny, primær malign svulst senere. Eksempelvis er det betydelig økt risiko for sarkom hos pasienter som er helbredet for genetisk betinget retinoblastom, og risikoen er ytterligere økt dersom pasienten har fått ekstern strålebehandling. Barn som har gjennomgått arvelig retinoblastom, skal derfor ha en annen oppfølging enn barn som har gjennomgått ikke- arvelig retinoblastom
- Ved genetisk årsak til kreft kan andre familiemedlemmer ha høy risiko for å bli syke (eksempel: Høy risiko for søsken ved arvelig retinoblastom der en av foreldrene har hatt retinoblastom)
Ved genetisk årsak til kreft med høy risiko for affisert barn vil noen foreldre ønske genteknologisk diagnostikk i nytt svangerskap (eksempel: Preimplantasjonsdiagnostikk ved arvelig retinoblastom)
De fleste former for arvelig kreft i barnealder følger dominant arvegang, evt. med nedsatt penetrans, men nær sagt alle arvemåter forekommer. I de fleste tilfeller med ett affisert barn foreligger en ny mutasjon. Gjentakelsesrisiko for søsken vil være lav, men gjentakelsesrisiko for barnets barn når det selv blir voksent og får egen familie, vil være opptil 50 %. Recessiv arv forekommer imidlertid (25 % gjentakelsesrisiko for søsken, meget lav risiko for barnets barn). Man bør være spesielt oppmerksom på at foreldrene (heterozygotene) til det syke barnet ved enkelte recessivt nedarvete (kreft-) syndromer selv har økt kreftrisiko (eksempel: mødrene til barn med Fanconi-anemi der årsaken er BRCA2-mutasjon, har risiko for arvelig bryst-eggstokkreft).
Det er indikasjon for å henvise barnet/familien til regional medisinsk genetisk avdeling (Oslo, Bergen, Trondheim, Tromsø) for utredning og genetisk veiledning ved mistenkt eller påvist arvelig kreft.
For mange av de arvelige barnekreftformene er tilgrunnliggende gen/mutasjon nå kjent og gentesting tilgjengelig ved norsk eller utenlandsk laboratorium. En oppdatert oversikt over tilgjengelige gentester ved norske laboratorier finnes på internettet (Norsk portal for genetiske analyser, www.genetikkportalen.no). Behandlende lege kan rekvirere gentesting på det syke barnet, mens gentesting av friske slektninger krever omfattende genetisk veiledning hos spesialist (§ 5, Bioteknologiloven). Tilsvarende vil man kunne finne informasjon om tilgjengelige tester internasjonalt på www.orpha.net (EU-sponset nettsted) og www.genereviews.org (amerikansk nettsted sponset av National Institutes of Health). Begge de sistnevnte nettstedene inneholder også oppdatert informasjon om arvelige sykdommer inkludert arvelig kreft hos barn.
På grunn av den rivende utviklingen innen fagfeltet er det lite hensiktsmessig å omtale hver barnekreftform i detalj. De regionale genetiske avdelingene vil ha oppdatert kunnskap. I tillegg kan man søke etter oppdatert litteratur, særlig slik den er tilgjengelig via de to forrige referansene.
Når skal en mistenke arvelig kreft hos barn?
Et lite mindretall av nær sagt alle typer barnecancer er arvelig betinget. I dag kan dette mindretallet ofte ikke diagnostiseres, men dette vil antakelig endre seg raskt fordi gentesting for tilgrunnliggende mutasjoner antas å ville bli lettere tilgjengelig og økonomisk mulig også i mange tilfeller der kreftsvulsten opptrer hos et ellers friskt barn fra en frisk familie. Et eksempel er muligheten for å påvise PHOX2B mutasjon ved nevroblastom (uten at dette er ledd i Congenitt Centralt Hypoventilasjons Syndrom).
Ny genteknologi (dypsekvensering, Neste generasjons gensekvensering NGS, High Throughput Sequencing HTS) kan komme til å revolusjonere diagnostikken ved barnekreft. Teknologien er på vei inn i rutinediagnostikken. De mmedisinsk genetiske avdelingene vil ha oppdatert informasjon om dette. De første resultatene kan tyde på at langt flere tilfeller av barnekreft skyldes monogen arv med høy penetrans eller genetisk predisposisjon til kreft enn tidligere antatt, men det er uklart om de første forskningsresultatene blir bekreftet og om dette vil føre til endret klinisk praksis (Krepischi et al., 2014; Zhang et al., 2015). Fortsatt (2019) er det særlig ved enkelte sjeldne kreftformer (som f.eks retinoblastom), ved syndromer eller ved opphopning av kreft i familien behandlende lege bør overveie muligheten for at det skulle kunne foreligge arvelig kreft. Flere hundre slike syndromer med større eller mindre risiko for kreft i barnealder er beskrevet, og inndeling kan foretas på mange måter. Nær sagt alle kreftformer er beskrevet som del av slike syndromer. Hodgson og Maher (Hodgson & Maher, 1999) og Offits (Offit, 1998) bøker inneholder tabellariske oversikter over feltet, men oppdaterte eller komplette lister finnes knapt siden feltet er i så rask utvikling.
Behandlende lege bør særlig vurdere om det kan foreligge arvelig barnekreft ved forekomst av:
- Familie med en enkelt kreftform der denne svulsttypen er kjent å kunne være del av et dominant arvelig kreftsyndrom. Eksempler er retinoblastom og Wilms’ tumor.
- Familie med opphopning av kjent sammensetning av kreftsykdommer. Eksempel er Li Fraumeni syndrom («brystkreft-sarkom-syndrom») der det karakteristiske er sarkom hos minst et barn i familien og brystkreft hos unge, voksne kvinner. Andre svulstformer forekommer både i voksen- og barnealder. Syndromet skyldes mutasjoner i TP53-genet.
- Pasienter med syndromer der hovedproblemet initialt oppfattes å være en medfødt misdannelse/utviklingsavvik, men der man erfaringsmessig også har risiko for å få kreft som barn. Det finnes svært mange slike syndromer (Krepischi et al., 2014; Zhang et al., 2015) hvorav en av de vanligste er nevrofibromatose 1 (risiko for mange typer svulster inkludert nevrofibrosarkom), Gorlin syndrom (risiko for flere typer svulster inkludert medulloblastom og basalcellecarcinom) og Multippel Endokrin Neoplasi type 2 (medullær thyroideacancer, andre). En undergruppe av disse er en del overvekst-syndromer inkludert Beckwith-Wiedemann syndrom der mekanismen synes å inkludere imprinting. Kreftrisikoens størrelse og betydning varierer fra syndrom til syndrom. I noen tilfeller er den høy hos et barn som har relativt små øvrige helseproblemer, i andre er den forøket men tallmessig ganske beskjeden sett i forhold til barnets øvrige helseplager. Pediatric Cancer Working Group, American association for Cancer Research, arrangerte høsten 2016 en ekspertkonferanse der man diskuterte predisposisjon for og overvåkning av personer med økt risiko for kreft. Anbefalingene fra denne konferansen er publisert og kan brukes som et utgangspunkt for overvåking av spesielle grupper kreftdisponerte individer1.
- Syndromer som kan identifiseres i barnealder, men gir kreft hos voksne. Eksempel: Familiær Adenomatøs Polypose (colon-polypper fra ungdomsalder, bl.a. colorectal cancer hos voksne), von Hippel Lindau syndrom (øyeforandringer hos barn, bl.a. nyrecancer hos voksne).
- Homozygote barn i familier med mutasjoner i BRCA2 eller MMR-gener (mismatch reparasjonsgener). I disse familiene er foreldrene (heterozygotene) disponert for hhv. arvelig bryst(-eggstokk)-kreft og Lynch syndrom (colorectal cancer, uterincancer, andre) i voksen alder. Barna får hhv. Fanconi-anemi type D1 (syndrom inkludert kreftrisiko) og er disponert for hematologiske maligniteter. Ofte blir familien identifisert gjennom det affiserte barnet som i tillegg til kreftrisikoen har pigmentforandringer i huden av café-au-lait type («nevrofibromatose-lignende bilde» klinisk).
- Familier med to barn med kreft, ett barn med to uavhengige kreftsvulster, bilaterale svulster i paret organ samt barn med svulster som kommer i en for svulsten uvanlig tidlig alder.
1Se https://www.aacr.org/ og http://clincancerres.aacrjournals.org/content/23/11
Molekylærgenetiske forhold
Sist faglig oppdatert: 26.05.2020
Basalbiologisk forskning har påvist en rekke gener som styrer celledeling, celledød, regulering av cellesyklus, evne til å reparere skadet DNA og effekt av medikamenter. Forandringer i disse gener kan føre til forandret vekstmønster med det resultat at en celle utvikles til en kreftcelle. Mange av disse forandringene er først og fremst funnet i kreftceller hos barn, og de brukes også diagnostisk for å skille de enkelte kreftformer når disse ikke kan defineres ved morfologi.
Flere barnekreftformer har derfor fungert som lyskastersykdommer i forståelsen av utvikling av kreft. Mutasjon i gener som hemmer vekst («tumor suppressor genes») kan være medvirkende til at cellevekst kommer ut av kontroll. Mest kjent er mutasjon i retinoblastomgenet (RB1), Ewing’s sarkom gen (EWS) og i Wilms’ tumor-genet (WT1). Mutasjon i RB1 kan være både somatisk og i kimbanen, det siste disponerer for multifokalt retinoblastom og andre kreftsykdommer som ben- og bindevevskreft. Økt antall av gener som fremmer vekst, kan fungere som onkogen. Mest utforsket er MYC-n genet ved nevroblastom der økt antall (amplifisering) av genet i kreftcellen gir en svært dårlig prognose.
I tillegg er det påvist translokasjoner og tap av genmateriale ved flere kreftformer. Kunnskap om disse forandringene bidrar i økende grad til å kunne skreddersy behandlingen (risikostratifisere behandlingen, se de enkelte sykdommer).
Om ny genteknologi
Ny genteknologi for å oppdage kopitallsavvik (array comparativ hybridisering aCGH) og avvik på nukleotidnivå (dypsekvensering, Neste generasjons gensekvensering NGS, High Throughput Sequencing HTS) er i ferd med å revolusjonere diagnostikken også ved barnekreft. Det gis derfor en kortfattet oversikt over relevante metoder. Gentesting kan gjøres i blod og/eller i tumorvev. Dette avsnittet omhandler gentesting i blod.
De aller fleste genetiske sykdommer innenfor arvelig barnekreft skyldes mutasjoner i enkeltgener (i motsetning til kromosomfeil). Man kan imidlertid mistenke kromosomfeil der pasienten i tillegg til kreftsykdommen har andre fenomener (som mental retardasjon eller alvorlige utviklingsavvik). Man kan da rekvirere array comparativ hybridisering (aCGH). Ved mistanke om enkeltgensykdom, rekvireres enten Sangersekvensering og eventuelt MLPA (Multiple Ligation Probe Assay) eller Neste Generasjons Sekvensering (NGS).
aCGH kan betraktes som en moderne, høykvalitets kromosomanalyse (selv om analysen gjøres på DNA nivå). aCGH oppdager kopitallsavvik. DNA isolert fra pasienten og fra en referansekilde (normalperson) merkes med ulike fluorokromer (typisk rød og grønn), denatureres (gjøres enkelttrådig), kappes opp i korte sekvenser og hybridiseres til et sett med komplementære DNA-sekvenser som er festet på en glassplate. I dag (2015) inneholder en plate til bruk i diagnostikken typisk 180,000 eller flere komplementære gensekvenser («prober»). Disse er arrangert i små «spots» på glassplaten. DNA fra pasienten og kontrollen vil feste seg på probene som har komplementær DNA sekvens. Reaksjonen gjøres kvantitativ slik at mengden rødt og grønt vil variere med hvor mye DNA som kommer fra pasientprøven og hvor mye som kommer fra kontrollen. Etter den kjemiske reaksjonen scanner man slide og registrerer hvilken farge hver enkelt spot har. Resultatet fores inn i en datamaskin som konstruerer et kart over kromosomene der det fremgår om det foreligger en, to eller tre kopier for hvert lite område av kromosomet. På denne måten kan man oppdage små kromosomområder som mangler en kopi (delesjoner) eller har tre kopier (duplikasjoner). Oppløseligheten ved lysmikroskopisk kromosomanalyse regnes å være slik at man kan oppdage avvik på rundt 5 million basepar (megabaser) mens man ved array CGH typisk kan oppdage avvik på 50,000 basepar.
Ved Sangersekvensering analyseres baserekkefølgen på enkeltgennivå i ett og ett gen. Det er bare den kodende delen (exonene og nærliggende del av intron) som blir analysert. Gjennomsnittlig finnes 85 % av mutasjonene i exons. For noen gener finnes erfaringsmessig (nær) 100 % av mutasjonene i exons, men generelt vil ikke en Sangersekvensering, eller noen annen genetisk laboratorieanalyse, kunne utelukke mutasjon i genet. Det vil oftest fremgå av litteraturen hvilken prosentandel av «typiske» pasienter man finner en mutasjon hos (typisk 85–95 %). Sangersekvensering vil ikke oppdage kopitallsavvik. For de vanligste genetiske sykdommene er under 10 % av mutasjonene delesjoner eller duplikasjoner, men det finnes mange sjeldne sykdommer der andelen er langt høyere (eksempel: ved von Hippel Lindau oppgis 28 % av mutasjonene å være kopitallsavvik). For de fleste genene vil det derfor være riktig også å undersøke for denne typen mutasjon. Den vanligste metoden å oppdage delesjoner og duplikasjoner i enkeltgener kalles MLPA (Multiplex Ligation-dependent Probe Amplification). Nærmere tekniske detaljer kan finnes for eksempel på [https://www.mrcholland.com/]. Metoden er rask med svar i laboratoriet på vel ett døgn. Den er enkel, billig og sensitiv og har mange anvendelsesområder. MLPA er basert på en multiplex PCR-reaksjon som kan undersøke opptil 50 ulike DNA segmenter samtidig. Det finnes over 300 probesett kommersielt tilgjengelig.
Ved Neste Generasjons Sekvensering kan man analysere mange (i prinsippet alle 21,000) genene samtidig. Analysemaskinene er installert på de genetiske avdelingene (og en del patologilaboratorier) også i Norge (2015). NGS kan utføres på mange måter og nye applikasjoner utvikles stadig. Per dato kan man betrakte NGS som en «multi-Sangersekvenseringsmetode». Teknologien vil antakelig også erstatte kopitallsanalyse (MLPA og aCGH) om få år. Analysen starter fra blod eller annet vev med rensing av DNA, deretter fragmenteres DNA, og man fisker ut de bitene av DNA som man ønsker analysert videre («capture»). Blant de mest aktuelle kommersielle capture kits i dag finnes de som fisker ut alle exoner (gir grunnlag for exom-sekvensering) eller alle exonene til alle gener som er kjent å ha sykdomsgivende mutasjoner (for alle mulige genetiske sykdommer). Det finnes også spesialdesignete kits for enkeltsykdommer/sykdomsgrupper, men capture kit for «alle sykdommer og syndromer som kan være forbundet med barnekreft» er ikke tilgjengelig i Norge idag. Etter prøveprepareringen, sekvenseres materialet i en maskin. Resultatet er en datafil med et sett med noen millioner korte sekvenser som settes sammen til stor gensekvens av et dataprogram (alignment). Resultat-filen analyseres ved at man filtrerer på de sekvensene man ønsker å se nærmere på. Dette gjøres enten ved at man velger aktuelle gener («filtrere en genliste») eller velger aktuelle gener ut fra arvemåte. Det første er antakelig foreløpig mest aktuelt ved barnekreft. Referanse (Zhang et al., 2015) er et godt eksempel på en svært omfattende genliste (over 500 gener), men en kan også lage kortere genlister for eksempel der den aktuelle krefttypen er kjent å kunne være forårsaket av et mindre antall gener (eks: sarcom hos et barn med et ukjent syndrom). Slike genlister er foreløpig ikke satt opp ved de genetiske laboratoriene i Norge.
Ved gentesting påviser man i prinsippet variasjon i DNA. Denne kan være sykdomsårsaken («patogen mutasjon») eller representere normal DNA-variasjon. Laboratoriene skal i prinsippet bare rapportere patogene mutasjoner og evt. varianter av ukjent betydning (Variant of Uncertain Significance = VUS) til kliniker. Antall påviste varianter stiger erfaringsmessig med antall gener analysert og vil være mye større ved NGS enn ved enkeltgentesting. Tolkningen av varianter lettes betydelig ved å se på prøver fra foreldrene: en VUS som også er til stede hos en frisk forelder vil vanligvis representere normalvariasjon. Foreldreprøver bør derfor sendes med prøver til NGS hvis mulig. Molekylærgenetiske funn (og patologifunn, andre funn) i tumorvev kan også brukes i tolkningen av variantene man har påvist i blod.
I barneonkologi er det i dag mest aktuelt å bruke Sangersekvensering med MLPA for å finne den genetiske sykdomsårsaken der pasienten har en tilstand der det bare er ett til to aktuelle gener (eks: retinoblastom, von Hippel Lindau syndrom) og NGS med filtrering på en liste gener (evt pluss MLPA) der pasientens tilstand kan skyldes mutasjon i ett av flere gener (eks Fanconi anemi). Ved undersøkelse av slektninger der man har funnet en mutasjon hos indekspersonen i familien, brukes Sangersekvensering eller MPLA.
Ved Sangersekvensering og MLPA kan man typisk vente svar fra laboratoriet etter 3–5 uker, ved Neste generasjons sekvensering 3–5 måneder. Svartidene forventes å bli kortere.
Behandling
Generelt om medikamentell behandling
Sist faglig oppdatert: 26.05.2020
Ser man bort fra CNS-svulstene der nær halvparten kan behandles med kirurgi alene, vil de aller fleste krefttypene hos barn kreve kjemoterapi. Kjemoterapien er detaljert beskrevet i de enkelte behandlingsprotokollene. Her nevnes kun noen få punkter med spesiell betydning for barn.
Cellegiftbehandling hos barn foregår praktisk talt utelukkende via sentralvenøs tilgang (sentralt venekateter eller veneport). Dette både pga større sikkerhet, men ikke minst for å spare barna for smerter og engstelse for stikk.
Barn er i vekst og utvikling, og visse bivirkninger av cytostatika som kan ha negativ innvirkning på dette, må begrenses. Dette gjelder spesielt:
- Kardiotoksisitet av antracykliner. De siste årene har den kumulative antracyklindosen i behandlingsprotokoller for barn blitt redusert så langt dette er forsvarlig. Protokollene foreskriver kardiologiske kontroller på spesifiserte tidspunkter. Disse er spesielt viktige dersom den kumulative dosen overstiger 200 mg/m2, men kardiotoksisitet kan også oppstå ved lavere kumulative doser enn dette.
- Nyretoksisitet av cisplatin, carboplatin, ifosfamid: Bruk av disse preparatene hos barn krever nøye oppfølging av nyrefunksjonen (GFR). I tillegg kan det utvikles en tubulær skade (renalt Fanconis syndrom) med betydelig tap av elektrolytter og bikarbonat i urinen.
- Ototoksisitet. Dette gjelder særlig cisplatin, i mindre grad carboplatin. Kombinasjon av disse medikamentene med ototoksiske antibiotika (aminoglykosider) må unngås. Vi fraråder derfor aminoglykosider i hele behandlingsforløpet hos barn som får platinholdig kjemoterapi. Det er viktig med audiometrikontroller underveis i behandlingsforløpet.
- Ernæringsproblemer på bakgrunn av kvalme, nedsatt matlyst, munnsår, generell nedsatt trivsel. Tidlig intervensjon med dietetiske tiltak, sondeernæring og/eller parenteral ernæring er nødvendig for å unngå negative virkninger for barnets utvikling.
For nærmere beskrivelse av cellegiftbehandlingen henvises for øvrig til kapitlene om de enkelte krefttyper. Mer generell informasjon vedrørende cytostatikabehandling finnes ellers i «Cytostatikaboken» (Medikamentell kreftbehandling: cytostatikaboken, 2009).
Kirurgi
Sist faglig oppdatert: 26.05.2020
Kirurgisk behandling av solide svulster utenfor sentralnervesystemet hos barn
Behandling av solide, maligne svulster hos barn er multimodal, og samarbeid i et tverrfaglig barneonkologisk team er viktig. For nesten alle solide svulster er kirurgi et sentralt ledd i behandlingen. Mange av svulstene som oppstår hos barn, er mer sensitive for kjemoterapi og stråleterapi enn maligne svulster hos voksne. Barn er ikke bare små voksne. Mens de vanligste kreftformer hos voksne oppstår i bryst, colon og prostata, er de vanligste solide svulstene hos barn (utenfor CNS) nevroblastom, nefroblastom og sarkomer. Terapiformer som er vanlige hos voksne, kan ofte ikke benyttes på samme måte hos barn. Fordi biologien i barnesvulster er annerledes, gir det unike muligheter for terapi, er med på å bestemme når det operative inngrepet skal skje i behandlingskjeden, og hvor omfattende kirurgien skal være. Det er derfor viktig at kirurgen blir involvert i planlegging av behandling og utredning av pasientene helt fra starten.
De aller fleste barn med maligne, solide svulster, behandles etter internasjonale behandlingsprotokoller. De som opererer barna, må være kjent med disse protokollene, være en del av et tverrfaglig barneonkologisk team, og kirurgien av solide svulster bør utføres av kirurger med solid erfaring med svulstkirurgi på barn.
Anbefalinger:
- Barn med maligne, solide svulster behandles etter internasjonale behandlingsprotokoller hvor kirurgi er en viktig intervensjon.
- Kirurgi av solide svulster på barn bør utføres av kirurger med solid erfaring med svulstkirurgi hos barn.
Kirurgisk behandling av svulster i sentralnervesystemet
Primærbehandlingen ved hjernesvulst hos barn er med få unntak kirurgisk fjerning av svulstvevet. Hos noen typer vil det være kurativt (B.J. Due-Tønnessen, Lundar, Egge, & Scheie, 2013; Lundar, Due-Tønnessen, et al., 2014). Langtidsoverlevelsen er også gunstigst for svulster som krever tilleggsbehandling hvis det ikke foreligger rester elle kun minimale rester av svulstvevet etter inngrepets avslutning.
Total svulstfjerning er dessverre ikke alltid mulig, men et kirurgisk inngrep vil uansett redusere svulstvolumet (bedre plassforholdene og derved bedre trykket i hjerneskallen), det gir en histologisk diagnose og i mange tilfeller kan man også gjenopprette hjernevæskesirkulasjonen (behandle vannhode/hydrocephalus) eventuell hydrocephalus.
Under det kirurgiske inngrepet utføres rutinemessig en MR undersøkelse (intraoperativ)for å vurdere det kirurgiske resultatet. Undersøkelsen kan vise gjenværende svulstvev som bør fjernes for et optimalt kirurgisk resultat og man vil da hvis hensiktsmessig fortsette inngrepet på bakgrunn av undersøkelsen.
Antall hjernesvulster hos barn er relativt lite sammenholdt med hjernesvulster hos voksne. Det er derfor vesentlig at ikke bare kirurgi, men også eventuell barne-intensiv behandling foregår i et team med bred erfaring tilpasset de spesielle utfordringer barnenevrokirurgiske inngrep krever.
Anbefalinger:
- Kirurgi er primærbehandling ved hjernesvulst hos barn. Komplett eller nær komplett reseksjon gir bedret overlevelse hos de fleste svulstgruppene. Det gjelder også hos de gruppene som er avhengig av tilleggsbehandling i form av stråle- eller medikamentell behandling for (langtids-)overlevelse (B.J. Due-Tønnessen et al., 2013; Lote et al., 2000).
- Peroperativ MR-undersøkelser kartlegger grad av reseksjon og vil kunne forbedre reseksjonsgraden.
- Kirurgi av hjernesvulster hos barn må utføres i omgivelser med solid erfaring innen avansert kirurgi og intensivbehandling på små barn.
Strålebehandling
Sist faglig oppdatert: 26.05.2020
Den tekniske forberedelse og gjennomføring av strålebehandling hos barn skiller seg lite fra strålebehandling hos voksne. Barnet må først immobiliseres i hensiktsmessig stilling. Det må deretter tas en CT av tumorområdet og evt andre risikoområder som skal bestråles, for å lage en doseplan. Kliniske data brukes, og diagnostiske bilder (CT, MR, PET) fusjoneres med doseplan-CT for å tegne inn svulsten i doseplan-CT-bildene. Stråleterapeuter bruker deretter avansert datagrafikk i et doseplansystem for å utarbeide hensiktsmessige strålefelter og stråledoser som vurderes og godkjennes av stråleonkolog. Deretter kan den planlagte behandlingen gjennomføres på stråleapparatet (oftest en lineærakselerator). Mindre barn som ikke kan klare å ligge stille på stråleapparatet, må ha anestesi for å kunne gjennomføre behandlingen. Dette kan bety opptil 30 seanser med anestesi i løpet av stråleperioden.
Strålebehandling er en effektiv lokalbehandling ved mange ulike typer kreft i barnealder. Normalvevet som omgir kreftsvulster hos barn, tåler imidlertid bare begrensede stråledoser før barnet risikerer å utvikle seinskader som etter behandling med høye stråledoser kan bli alvorlige. All strålebehandling hos barn blir da en vanskelig avveining mellom risikoen for å dø av kreftsykdommen og risikoen for å utvikle livslange seinskader etter livreddende strålebehandling. En slik risikoavveining reiser etiske problemer for foreldre og behandlere: Hvor store skader kan man akseptere på barnets vegne før prisen for å overleve blir for høy? Retningslinjer gitt i internasjonale behandlingsprotokoller for strålebehandling ved barnekreft er en god støtte i disse vanskelige situasjonene.
Seinskadene består av veksthemning i det bestrålte vevet, nedsatt funksjon av bestrålte organer og en moderat, men trolig livslang overrisiko for å utvikle nye godartede eller ondartede svulster mange år seinere (Oeffinger et al., 2006). Seinskadene vil ofte gradvis øke på utover i vekstperioden og blir derfor særlig uttalte hos de barna som er yngst på behandlingstidspunktet. Svært alvorlige kognitive skader må forventes etter høye stråledoser mot store deler av hjernen hos barn yngre enn 3 år, og strålebehandling har derfor ikke vært brukt for denne pasientgruppen. Med ny teknologi vil man kunne begrense stråledosen til normalvevet rundt svulsten bedre enn man kunne før, og dette vil etter hvert kunne medføre en viss endring av praksis. Intens cellegiftbehandling gir trolig mindre risiko for seinskader og kan i noen grad erstatte strålebehandling hos de minste barna.
For større barn og tenåringer avtar risikoen for seinskader med stigende alder. Toleransen for stråling hos barn avhenger av en rekke faktorer: Barnets alder, hvilket organ som bestråles, størrelsen på vevsvolumet som bestråles, stråledosen gitt per dag (fraksjonsdosen), og total stråledose er de viktigste.
Selv om strålebehandling gir risiko for langtidsskader, gir slik behandling et viktig bidrag til å bedre sjansene for langtidsoverlevelse ved kreftsykdommer som medulloblastom, manifeste CNS-leukemier, og intrakraniale germinomer. Tillegg av strålebehandling øker mulighetene for helbredelse ved en rekke andre maligne tilstander i barnealder (Hodgkins lymfom, ulike sarkomtyper, avanserte stadier av Wilms tumor og nevroblastom, samt ved lokaliserte carcinomer eller andre lokaliserte svulster), særlig dersom det ikke er mulig å gi lokalbehandling i form av kirurgi.
Konvensjonell strålebehandling gis i hovedsak med fotonbasert teknikk (røntgenstråler), i noen grad benyttes også elektronstråling. Det finnes også teknologi for partikkelstråling, for barn er det først og fremst protonbehandling som er aktuelt (Olsen, Bruland, Frykholm, & Norderhaug, 2007). Protonstrålingens største fordel sammenlignet med konvensjonell strålebehandling er at det er mulig å avgrense strålefeltet bedre enn ved fotonstråling. Av den grunn vil man kunne begrense stråledosen til friskt vev omkring tumor og derved redusere skadevirkningene som følge av strålebehandlingen. For barn som har en prinsipielt kurabel kreftsykdom er dette spesielt viktig; de skal leve med ettervirkningene av kreftbehandling i mange år. Det er derfor avgjørende at man i størst mulig grad forsøker å redusere stråledosen til normalt vev. Av denne grunn bør de fleste barn som skal motta strålebehandling vurderes for protonstråling. Viktigst er dette for de yngste barna, de som skal ha høye stråledoser, og de som skal strålebehandles i områder med mange strålesensitive organer i nærheten. Det siste vil spesielt gjelde svulster i sentralnervesystemet, hode/hals-regionen og bekkenet. Ved noen tumorgrupper, for eksempel lymfomer, behandler man pr i dag et risikovolum og det vil da være mindre aktuelt med protonbestråling.
Det er ingen holdepunkter for at protonstråling i seg selv er mer effektivt enn konvensjonell strålebehandling. Dog kan protonstråling på grunn av lavere doser til omkringliggende normalvev, gi muligheten til doseeskalering dersom det er ønskelig. Dette er nok mer aktuelt for voksne enn for barn. Protonstråling er ikke etablert i Norge og selv om det nå stadig etableres flere protonsentre verden over, så er tilgjengeligheten foreløpig noe begrenset. Imidlertid har man nå en nasjonal avtale, som håndteres av hver av de fire helseregionene, med flere utenlandske sentra for lettere å kunne tilby protonbehandling til norske pasienter. Barnekreftpasientene er blant de som prioriteres høyest for dette. Det er god grunn til å tro at protonstråling vil bli vesentlig mer brukt i fremtiden.
Anbefalinger:
- All strålebehandling hos barn er en avveining mellom risiko for sykdomsprogresjon og senskader av stråling. Spesielt hos barn under 3 år med hjernesvulst skal det med bakgrunn i dette nøye vurderes om det er aktuelt med strålebehandling.
- Internasjonale behandlingsprotokoller for strålebehandling ved barnekreft vil være veiledende for hvilke pasientgrupper som tilbys strålebehandling og hvilke stråledoser som gis.
- CT av området som skal bestråles må tas for å utarbeide en doseplan.
- Mindre barn vil ofte trenge anestesi under strålebehandlingen.
- Protonstrålebehandling gir mindre dose til normalvev og vil derved redusere skadevirkningene etter strålebehandling. Man mener derfor at spesielt barn har fordeler av slik behandling. Protonbehandling er ikke etablert i Norge, men det er nylig gjort avtaler med utenlandske sentra for å kunne tilby slik behandling til norske pasienter. De fleste barn som trenger strålebehandling og som kan kureres for sin kreftsykdom, bør vurderes for protonbehandling utenlands. Spesielt gjelder dette de yngste barna, barn som trenger høye stråledoser og barn med svulster i nærheten av strålesensitive organer.
Utprøvende behandling
Sist faglig oppdatert: 26.05.2020
Behandling av kreft hos barn har vist store fremskritt de siste 40 år. Hovedgrunnen til denne fremgang er internasjonalt samarbeid og kliniske forskningsprotokoller. En viktig del har vært den utprøvende behandling der også fagmiljøene i Norge deltar. Utprøvende behandling vil derfor være en viktig faktor for fortsatt fremgang. Nye aktuelle protokoller blir diskutert i Kompetansetjenestens faggrupper som anbefaler nasjonale retningslinjer for 1. linjebehandling.
Når fagmiljøet går inn for en eksperimentell behandling (Fase 1/2 studie) som kan gis i et land i Norden er det lagt føringer for at dette skal dekkes av de regionale helseforetak (enhet for utenlandsbehandling), etter behandling av dette spørsmålet (på bakgrunn av henvendelse fra barneonkologimiljøet) på det interregionale fagdirektørmøtet 14.12.2015. Dette gjelder kun behandling av barn som kan utføres i Norden, og som er vurdert hensiktsmessig (inkl. kost/nyttevurdert) av behandlende lege. En oversikt over aktuelle eksperimentelle protokoller som er åpen for inklusjon ligger på NOPHOs nettside https://www.nopho.net/ (under «Protocols», novel therapies), og vil dessuten være tilgjengelige på www.clinicaltrials.gov.
Akutte onkologiske tilstander
Sist faglig oppdatert: 26.05.2020
I sjeldne tilfeller debuterer kreftsykdommen med dramatiske, livstruende symptomer, men mer vanlig er det at barnet under behandlingen utvikler det som betegnes akutte onkologiske tilstander. Slike tilfeller krever rask sykehusinnleggelse på barneonkologisk eller intensiv avdeling. Det anbefales å konferere med barneonkolog (dersom mulig) om behov for behandling før og under transport. I det følgende gjennomgås kort de hyppigste tilstander som ses ved diagnosetidspunkt og under behandlingen. Anbefalinger for behandling av akutte onkologiske tilstander finnes beskrevet i Barnelegeforeningens Akuttveileder (Akuttveileder i pediatri [e-bok], 2013).
Superior vena cava syndrom (SVCS) og superior mediastinalt syndrom (SMS)
Store tumormasser kan medføre obstruksjon av vitale organer som luftveiene, sentrale kar eller spinalkanalen. Ved superior vena cava syndrom (SVCS) og øvre mediastinalt syndrom (SMS) komprimeres henholdsvis vena cava og trachea, eventuelt høyre hovedbronkus av tumormasser i mediastinum. Ved diagnosetidspunktet forekommer dette hyppigst ved non-Hodgkins lymfom, Hodgkins lymfom, T-celle akutt lymfatisk leukemi, germinalcelletumores og sjeldnere ved nevroblastom og Ewings sarkom. Senere i behandlingsforløpet vil SVCS oftest oppstå på grunn av tromber. Klinisk vil barnet være preget av dyspne, hoste og hvesing, svelgvansker, brystsmerter, hevelse i ansikt, hals og overkropp og struttende vener i samme område. Hvis tilstanden progredierer, ses tiltagende engstelse, nedsatt bevissthet, synsforstyrrelser, kramper og synkope.
Observasjon og behandling bør foregå på intensivavdeling med mulighet for rask intubering og respiratorbehandling. Behandling må ofte prioriteres foran diagnostikk, da utredning med generell anestesi eller sedering kan forverre den kliniske tilstand. Primærutredning begrenses til klinisk undersøkelse, røntgen av thorax, laboratorieprøver og eventuelt BM og biopsi i lokalanestesi. Hvis anestesi eller sedering vurderes, må en grundig risikovurdering med CT, ekkokardiografi og respiratorisk evaluering i sittende og liggende stilling gjennomføres. I de fleste tilfeller inntrer en betydelig bedring 12–24 timer etter at behandling med cytostatika eller mediastinal bestråling er startet. Behandlingen er dog ikke uten risiko da bestråling kan utløse et lokalt ødem med ytterligere kompresjon av vitale strukturer. Ved transport til en barneonkologisk avdeling er det viktig at pasienten ikke legges flatt, men transporteres i mest mulig sittende stilling, for å unngå luftveiskollaps.
Medulla spinalis kompresjon
Hos enkelte barn kan de første symptomene være gangvansker, svakhet i armer eller ben som tegn på påvirkning av ryggmargen. Tilstanden kan inntreffe i løpet av få dager med ryggsmerter, lokalt eller utstrålende eventuelt med påvirkning av blære og endetarmsfunksjon og senere lammelser.
Hyppigste årsak er sarkomer, nevroblastomer, lymfomer og leukemi, men også spredning fra svulster i hjernen eller svulster i ryggmargsvevet
Under eller etter behandling kan skjelettet i ryggsøylen bli påvirket (benskjørhet) eller behandlingsrelatert direkte påvirkning av nervevevet i ryggmargen (myelitt). Det er svært viktig at tilstanden utredes uten tidstap (MR undersøkelse) og at nevrokirurg (og evt. ryggortoped) kontaktes uten opphold. Behandling bør igangsettes raskt med høy dose steroider, deretter bestråling, cytostatika eller kirurgi, avhengig av etiologi og symptomer, basert på tverrfaglig diskusjon av hvert enkelt tilfelle.
Økt intrakranielt trykk ved tumor cerebri
Hjernesvulster kan i løpet av kort tid gi et forhøyet hjernetrykk. Forløpet kan være svært dramatisk og kan føre til raskt bevissthetsendring og i verste fall død.
De dramatiske symptomene kan skyldes svulstvevets raske vekst, spontane blødninger i svulstvevet eller at svulstvevet hindrer hjernevæskens naturlige sirkulasjonsveier og fører til akutt vannhode/hydrocefalus enten ved at en av de naturlige vannveiene blokkeres, men også ved en diffus blokkade av evnen til å ta opp hjernevæske som vi ser ved en diffus spredning av sykdommen (generalisert carcinomatose) langs hjernehinner og blodårer (vener).
Tidligst mulig diagnose (før svulstvevet gir et forhøyet hjernetrykk) er derfor viktig for å unngå en vanskelig kontrollerbar situasjon med raskt økende symptomer/bevissthetstap. Ved mistanke om svulst i hjerne eller ryggmarg / forhøyet hjernetrykk må det utføres en MR undersøkelse av hjerne og om mulig også ryggmarg. Denne undersøkelsen vil i de fleste tilfeller være avklarende etter dialog/konferering med barneonkolog og nevrokirurg. I en situasjon med mistanke om økende symptomer og ikke mulighet for MR undersøkelse vil en CT-undersøkelse også kunne avklare alvorlighetsgrad i en akutt situasjon før overflytning til en nevrokirurgisk eller regional barnemedisinsk avdeling.
Symptomer på et forhøyet hjernetrykk avhenger særlig av alder og lokalisasjon av tumor. Typisk dreier det seg om morgenhodepine og oppkast, generell ustøhet, eventuelle personlighetsforandringer, synsforstyrrelser og nakkestivhet.
Hos små barn med åpen fontanelle vil et svært raskt økende hjernetrykk påvises ved en spent bulende fontanelle, ved en mer langsom trykkøkning vil man observere en progressivt økende hodeomkrets.
Begge situasjoner må utredes raskt, se pakkeforløp, kreft hos barn.
Hos større barn påvirkes barnets våkenhet og etter hvert som situasjonen forverres etter også respirasjonsmønster konferer undersøkelse av den komatøse pasient i akuttveileder.
Oftalmoskopi har en begrenset plass ved undersøkelse av en pasient med raskt innsettende symptomer og synkende bevissthet. Funn av stasepapiller hos en pasient med langvarige diffuse symptomer krever rask videre henvisning og utredning.
Sekundær hydrocephalus krever rask vurdering og i noen tilfeller akutt nevrokirurgisk intervensjon med drenasje og/eller behandling med steroider.
Epileptiske anfall i denne situasjonen må kuperes raskest mulig og intubasjon sikring av luftveier må vurderes før akutt overflytning til nevrokirurgisk evt. barneonkologisk avdeling. Tilstandender som gir mistake om et progressivt økende hjernetrykk / truende herniering krever umiddelbar nevrokirurgisk intervensjon. Det henvises også til retningslinjer i Barnelegeforeningens Akuttveileder (Akuttveileder i pediatri [e-bok], 2013).
Hyperleukocytose og leukostase
Hyperleukocytose er karakterisert ved leukocyttall over 100 x 109/L i perifert blod og forekommer oftest ved akutt lymfatisk og myelogen leukemi hos barn. Risiko for komplikasjoner er ikke bare avhengig av celletall, men også hvilken form for leukemi barnet har.
Ved akutt myelogen leukemi er myeloblastene og særlig monoblastene store og rigide, «klebrige» og sirkulerer dårlig gjennom mikrosirkulasjonen. Høyt celletall øker derfor risikoen for hyperviskositet og leukostase, karakterisert ved intravaskulær aggregering av blaster, karskade og obstruksjon, trombose og blødning. Hyppigst affiseres lunger og CNS. Symptomene er dyspne, hypoksi, fulminant respirasjonssvikt og eventuelt høyresidig ventrikkelsvikt. CNS-affeksjon er tilsvarende alvorlig, med utvikling av intrakraniell blødning eller trombose som hyppig dødsårsak. Priaprisme kan også ses hos barn. Behandling er reduksjon av viskositet med økt hydrering og begrenset bruk av blodtransfusjon (transfundert Hb bør ikke overstige 8 g/dl). Diuretika bør unngås i primærfasen, og barnet bør raskt overflyttes til barneonkologisk avdeling etter stabilisering. Hydrerings- og transfusjonsregimet må diskuteres med mottagende avdeling før transport, men aggressiv hydrering med minimum 3000 ml/m2/døgn er alltid indisert. K-tilskudd er sjelden nødvendig og vil kunne forverre tumorlysesyndrom. Tumorlyseprofylakse beskrives nedenfor. Etter overflytting fortsettes symptomatisk behandling og eventuelt leukaferese eller utskiftingstransfusjon for mindre barn. Rask start start av cytostatika/steroider er nødvendig. Ved akutt lymfatisk leukemi er tumorlyse ved behandlingsstart en viktigere risikofaktor enn leukostase. Dette skyldes at lymfoblastene er mindre og mer fleksible og oftest svært sensitive for cytostatika. Alvorlige komplikasjoner ses sjelden før leukocyttallet er over 400 x 109/L ved ALL. Det henvises også til retningslinjer i Barnelegeforeningens Akuttveileder (Akuttveileder i pediatri [e-bok], 2013).
Tumorlysesyndrom (TLS)
Tumorlysesyndrom er en potensielt livstruende tilstand med hyperurikemi, hyperkalemi, hyperfosfatemi og sekundær utvikling av nyresvikt og hypokalsemi. TLS kan oppstå før start av kjemoterapi (etter oppstart hydrering), men ses oftest etter 1 til 5 dager, hvor store cellemasser nedbrytes over kort tid, og ekskresjonskapasitet for metabolitter overskrides. Tilstanden ses hyppigst ved leukemier (spesielt T-celle ALL), T-celle lymfom, Burkitt lymfom men også ved andre lymfomer og akutt myelogen leukemi. Strålebehandling i akutte situasjoner kan også utløse TLS. Forebyggende behandling igangsettes før overflytting til barneonkologisk avdeling ved mistanke om store tumormasser og rask celledeling. Viktigste behandling består av forsert diurese og væsketerapi med 3000–4500 mL/m2 per døgn, og det er sjelden indikasjon for K-tilskudd (i hydreringsvæsken) i akuttfasen. Ved manifest tumorlyse eller tilstedeværelse av risikofaktorer (se ref (Akuttveileder i pediatri [e-bok], 2013) for risikogruppering) gis rasburicase (Fasturtec®) i.v.; en svært effektiv urat-oksidase. NB ved måling av urat under Fasturtec-behandling, må prøven transporteres på is til laboratoriet (ellers vil nedbrytingen fortsette ex vivo og svaret være falskt lavt). Ved mindre risiko for TLS kan Allopurinol per os gis.
Før overflytting av pasient til videre utredning og behandling bør man konsultere barneonkologisk avdeling om valg av forebyggende behandling. Senere, når steroid- eller cytostatikabehandling er startet, må barnet monitoreres for utvikling av elektrolyttforstyrrelser og tegn til nyresvikt. I enkelte tilfeller blir det behov for hemodialyse, og nefrolog bør konsulteres tidlig i forløpet. For ulike risikogrupper og detaljer vedrørende behandling, se Barnelegeforeningens Akuttveileder (Akuttveileder i pediatri [e-bok], 2013).
Andre akutte onkologiske tilstander
Blant andre akutte onkologiske tilstander kan nevnes koagulopati og DIC, spesielt ved akutt myelogen leukemi type M3 og M5, «All-Trans retinoic acid syndrom» ved behandling av promyelocyttleukemi, hyperkalsemi (sjelden hos barn, men kan oppstå ved osteolytiske beintumores, immobilisering og andre solide svulster og leukemier), samt posteriort reversibelt encefalopati syndrom (PRES), spesielt i startfasen av leukemi/non-Hodgkin lymfom behandling. Endelig skal nevnes SIADH (Syndrome of Inappropriate ADH Secretion) som forekommer ved hjernesvulster, lymfom og leukemier, eventuelt ved bruk av cyklofosfamid og vinkristin. Tilstanden skyldes ukontrollert utskillelse av ADH som medfører vannretensjon og hyponatremi. Behandling er væskerestriksjon i lette tilfeller og langsom normalisering av elektrolyttforstyrrelsene.
Under behandlingen for kreft ses alvorlige akutte tilstander som typhlitt, pancreatitt og venookklusiv leversykdom. Sistnevnte oppstår ved celleskade og sekundær trombosering av de små levervenene. Det henvises her til spesiallitteratur for detaljert informasjon.
Behandling av de enkelte kreftsykdommer
Leukemier, lymfomer og histiocytoser
Sist faglig oppdatert: 26.05.2020
Leukemier
Det diagnostiseres hvert år ca. 35–40 nye tilfeller av leukemi hos barn under 15 år i Norge (Nasjonalt kvalitetsregister for barnekreft, 2015). De fleste, 80–85 %, er akutt lymfatisk leukemi (ALL), 10–15 % er akutt myelogen leukemi (AML), mens et fåtall utgjøres av andre, sjeldne leukemityper («voksen» type kronisk myelogen leukemi – KML med Philadelphia-kromosom, juvenil myelomonocyttleukemi – JMML). Heldigvis har prognosen for de vanligste leukemiformene bedret seg mye i løpet av de siste 30 årene, slik at majoriteten av barna kan regne med å bli helt friske (Forestier & Schmiegelow, 2006; G. Gustafsson et al., 2000; Kolmannskog et al., 2007; Pui, 2006; Pui & Evans, 2006; Pui, Schrappe, Ribeiro, & Niemeyer, 2004; Schmiegelow et al., 2010).
Behandling av leukemi hos barn er i Norge et regionalt ansvar. Barn med symptomer og funn som gir mistanke om leukemi, skal straks henvises tiltrengende øyeblikkelig hjelp til lokal og senere regional barneavdeling til utredning og behandling. Ofte må det startes væskebehandling og annen støttebehandling før overflytting til regionsykehus, se avsnittet om akutte onkologiske tilstander ovenfor. Når leukemidiagnosen er verifisert, startes behandlingen ved regionsykehusets barneavdeling, mens den videre behandlingen skjer i et nært samarbeid mellom regional og lokal barneavdeling.
Diagnose
Symptomer som kan gi mistanke om leukemi, er vanligvis uspesifikke: blekhet, blåmerker, hovne lymfeknuter, feber, skjelettsmerter. Ved klinisk undersøkelse finner man ofte at barnet ser påfallende sykt ut, og mange har hovne lymfeknuter, tegn på økt blødningstendens (blåmerker, petekkier) og forstørret lever og milt. Noen har hovne ledd. Klinisk undersøkelse må suppleres med blodprøver som ofte vil avsløre anemi, nøytropeni og trombocytopeni. Mange barn blir innlagt i barneavdeling pga. en akutt infeksjon uten at leukemimistanken er reist på forhånd.
Diagnosen stilles ved benmargsundersøkelse, kun unntaksvis kan diagnosen stilles ved mikroskopi av blodutstryk alene. Mikroskopi av benmargsutstryk må suppleres med benmargsbiopsi, immunfenotyping av benmargsaspirat, benmargskaryotyping og et sett molekylærbiologiske undersøkelser som er definert i behandlingsprotokollene. Det må også utføres spinalpunksjon for å se om det er spredning av leukemiceller til sentralnervesystemet (CNS), men denne prosedyren utsettes til diagnosen er sikker nok til at man kan gi første cellegiftdose rett inn i spinalvæsken samtidig.
Behandling
Behandling av leukemi hos barn skjer etter felles nordiske retningslinjer vedtatt av NOPHO (NOPHO – nordisk forening for pediatrisk hematologi og onkologi https://www.nopho.net/) som er en sammenslutning av nordiske barneonkologer fra alle de fem nordiske landene. NOPHO har egne behandlingsprotokoller for ALL og AML hos barn, og NOPHO har som gruppe tilsluttet seg Interfant-protokollen til behandling av ALL hos spedbarn, EsPhALL-protokollen til behandling av ALL med Philadelphia-kromosom, ICC APL Study 01 til behandling av akutt promyelocyttleukemi, ML-DS 2007 studien til behandling av barn med myeloid leukemi ved Down syndrom og EWOG-MDS 2006 til behandling av JMML hos barn. Alle protokollene er tilgjengelige på den lukkede delen av NOPHOs nettsider.
ALL
ALL deles inn i risikogrupper på bakgrunn av definerte risikofaktorer som er til stede ved diagnosetidspunktet, kombinert med respons på tidlig behandling. Risikofaktorene er modernisert og definert ut fra forskningsresultater oppnådd i NOPHOs tidligere behandlingsprotokoller (Forestier & Schmiegelow, 2006; G. Gustafsson et al., 2000; Kolmannskog et al., 2007; Schmiegelow et al., 2010).
Faktorer det tas hensyn til ved risikostratifisering av ALL:
- Antall hvite blodlegemer
- Alder
- Immunfenotype (B-precursor vs. T)
- Cytogenetikk – Visse definerte molekylærbiologiske karakteristika
- Respons på behandling definert som antall gjenværende leukemiske blaster i benmargen ved definerte tidspunkter.
I NOPHO ALL-2008 protokollen var alder tatt ut som selvstendig risikokriterium, dette er tatt inn igjen i den nye ALLTogether (A2G) protokollen. Generelt legges det nå mer vekt på cytogenetiske og molekylærbiologiske egenskaper ved de leukemiske blastene, kombinert med tidlig behandlingsrespons vurdert ved MRD (MRD – minimal residual disease) undersøkt ved hjelp av flowcytometri (pre-B-ALL) eller PCR-undersøkelser.
Den følgende risikostratifiseringen gjelder for A2G protokollen: Det skilles mellom Standard risk (SR), IR-low, IR-high, HR-chemo og HR-SCT. Tabellen nedenfor gir en oppsummering (tatt fra ALLTogether pilot versjon 1.4). Inndelingen er komplisert og stratifisering må i hvert tilfelle gjøres omhyggelig ved hjelp av nyeste protokollversjon.
| Stratification | Stratification criteria | |
| Standard-Risk (SR) | No T-cell ALL No HR-genetics* + No MRD-signal day 29 (end of induction) No CNS3/TLP+ No ABL-class fusion | |
| IR | MRD <5% day 29 | |
| IR-low (IRL) | <16 years at diagnosis No HR-genetics+MRD response as specified below | |
| Sub-group | MRD cut-off | |
| ETV/RUNX1 | MRD d29 <0.1% | |
| High hyperdiploid | MRD d29 <0.03% | |
| B-other + CNA GR** | MRD d29 <0.05% | |
| T-cell | No MRD signal d71 | |
| TLP+ <5 WBC/microL CSF | No MRD signal d29 | |
| IR-high (IRH) | Sub-group | MRD cut-off |
| CNS3-involvement TLP+ ≥5 WBC/microL CSF | Any MRD <HR-criteria | |
| Poorly responding testicular/mediastinal disease | Any MRD <HR-criteria | |
| CNA PR*** | Any detectable MRD d29 <5% | |
| Genetic groups as above | MRD ≥ cut-off above | |
| Failed genetic work-up | Any MRD <HR-criteria | |
| Failed MRD work-up | All patients | |
| T-cell | If MRD d29 <5% MRD <0.05% d 71 If MRD d29 ≥5% MRD <0.5% d 50 and undetectable d71 | |
| ≥ 16 years | Not SR/HR-criteria | |
| ABL-class fusions | Any MRD d29 MRD< 0.05% d71 | |
| HR-chemo BCP | t(17;19) and no HSCT-indication | |
| <16 years and MRD ≥5% d29 but MRD <0.05% d71 | ||
| <16 years, NCI HR at diagnosis and MRD ≥0.01% at TP2 | ||
| <16 years and remaining testicular disease/med mass ≥1/3 of initial volume after Consolidation 1 | ||
| HR-HSCT BCP | <16 years and MRD ≥0.05% d71 | |
| <16 years MRD d 29 ≥5% and ≥0.5% day 50 | ||
| ≥16 years and any HR-criteria | ||
| HR-HSCT T-cell | MRD d29 <5% and ≥0.05% day 71 | |
| <16 years MRD d 29 ≥5% and ≥0.5% day 50 | ||
| ≥ 16 years MRD ≥5% day 29 | ||
| <16 years MRD d29 ≥5%, <05% day 50 and detectable day 71 | ||
| Remaining testicular disease/med mass ≥1/3 of initial volume after Consolidation 1 | ||
Behandlingen i A2G-protokollen er intensiv kombinert cytostatikabehandling som bygger på tidligere NOPHO protokoller. Behandlingen er inndelt i induksjonsfase, konsolideringsfaser, sen intensivering og vedlikehold for SR- og IR-gruppene, mens pasienter med HR-ALL får, istedenfor en vanlig vedlikeholdsfase, intensivert kjemoterapi, stamcelletransplantasjon eller CAR-T. Varigheten av behandlingen er 2 år fra avsluttet induksjonsbehandling for alle risikogrupper unntatt pasienter som får behandling med SCT eller CAR-T.
Generelt om ALLTogether (A2G) protokollen
Protokollen er en kombinert behandlings- og forskningsprotokoll. Inklusjon krever skriftlig informert samtykke fra foreldrene. Barn som behandles etter denne protokollen, skal registreres online i ALLTogetherdatabasen. Registrering skal skje innen 1 uke etter diagnose og deretter fortløpende, hvor spesielt registrering av «events» (tilbakefall, manglende behandlingsrespons, død i komplett remisjon, sekundær kreft) og toksisitet er viktig.
- Induksjonsfasen består av dexamethason, vinkristin, asparaginase, og intratekal behandling (medikamenter som settes direkte inn i spinalvæsken for å beskytte mot leukemi i CNS). Pasienter med T-celle ALL og andre risikokriterier får i tillegg daunorubicin.
- Første konsolideringsfasen er avhengig av risikogruppe (SR vs. høyere risiko) og består av nedtrappende dexamethason etterfulgt av peroral 6-merkaptopurin og 4 uker med lavdose cytarabin samt 2–3 doser asparaginase. I tillegg får de høyere risikogruppene cyklofosfamid og intratekal behandling
- Andre konsolideringsfaser er avhengige av risikogruppe og består bl.a. av asparaginase og høydose-metotrexat kurer. Antallet HD-MTX kurer er dog vesentlig redusert i denne protokollen sammenlignet med tidligere NOPHO protokoller. Foreløpig (i pilotfasen) får alle pasienter doxorubicin i sen-intensiveringen, senere vil SR pasienter som randomiseres til eksperimentell arm kunne få hele behandlingen uten doxorubicin og dermed uten antracykliner i det hele tatt.
- Vedlikeholdsbehandlingen inneholder 6-merkaptopurin og metotreksat i tablett- eller miksturform og for alle risikogrupper unntatt SR gjentatte pulser med vincristin/dexamethason.
Protokollen inneholder i pilotfasen ingen randomiseringer. Når master-protokollen åpner i løpet av 2020 vil det være 3 randomiseringer:
- Randomisering 1: Standard riskgruppe. I senintensivering randomiseres pasienter mellom doxorubicin (standard arm) og ikke doxorubicin (eksperimentell arm), kfr. ovenfor. Dette er en non-inferiority studie.
- Randomisering 2: Intermediate-low riskgruppe. Randomisering i 3 grupper: Standard arm: doxorubicin i senintensivering og vincristin/dexapulser i vedlikeholdsfasen. Eksperimentell arm 1: Ikke doxorubicin i senintensivering men vincristin/dexapulser i vedlikehold. Eksperimentell arm 2: Doxorubicin i senintensivering men ikke vincristin/dexapulser i vedlikehold.
- Randomisering 3: Intermediate-high riskgruppe. Randomisering av vedlikeholdsbehandlingen i 3 grupper: Standard arm: Vincristin/Dexa pulser hele veien til behandlingen avsluttes. Eksperimentell arm 1: Inotuzumab (eksperimentell terapi) i 6 uker, deretter VCR-dexa pulser som i standard arm. Eksperimentell arm 2: Som standard arm, men tillegg av 6-tioguanin (TEAM).
For hver randomisering kreves eget samtykke fra foreldrene.
Anbefalinger:
- Pasienter med ALL bør behandles etter den til enhver tid gjeldende NOPHO-protokollen, p.t. er dette A2G protokollen (pilot versjon) mens fullversjonen av A2G forventes åpnet i løpet av 2020. Det kreves eget samtykke fra foreldre.
- Et fåtall pasienter behandles med stamcelletransplantasjon eller CAR-T i første remisjon.
Spedbarns-ALL
Hos barn som er < 1 år ved diagnose, anbefaler NOPHO bruk av «Interfant 06»-protokollen, en videreutvikling av Interfant-99 protokollen, som er hittil beste publiserte behandling (Pieters et al., 2007) (Evidensgrad B). Protokollen ligner på intermediærrisikogruppen av den ordinære ALL-protokollen, men inneholder mer cytarabin, noe som anses som gunstig ved MLL-rearrangeringer som er til stede i de fleste tilfellene av spedbarns-ALL. Protokollen inneholdt tidligere en randomisering, hvor den eksperimentelle armen hadde 2 AML-lignende blokker, dette er avsluttet og man bruker standard arm. Protokollen brukes i Norge som «best available treatment», og man har fulgt standard arm hele tiden. Alle risikogrupper følger prinsipielt samme behandling, men pasienter i høyrisikogruppen og pasienter i intermediærgruppen med dårlig terapirespons tilbys i tillegg stamcelletransplantasjon (G. Mann et al., 2010) (Evidensgrad B).
Anbefalinger:
- Spedbarn med ALL bør behandles etter Interfant 06-protokollen, og følge standardarm. Det kreves eget samtykke fra foreldre. (Evidensgrad B)
- Pasienter i høyrisikogruppen og pasienter i intermediærgruppen med dårlig respons (definisjon, se protokoll) tilbys stamcelletransplantasjon. (Evidensgrad B)
Philadelphia-kromosom-positiv ALL
Ca 3 % av barn med ALL får påvist Philadelphia-kromosom (bcr-abl) i forbindelse med utredning for ALL. Siden dette svaret ikke er kjent fra starten, vil barna starte behandling etter NOPHO ALL 2008 ( evt A2G)-protokollen, men etter induksjonsfasen overføres de til behandling etter en internasjonal protokoll, EsPhALL, tilpasset nordiske forhold av NOPHO («EsPhALL amendement for NOPHO»). Det som skiller behandlingen av barn etter denne protokoll fra ALL-behandlingen for øvrig, er først og fremst behandling med imatinib (Glivec®) som gis fra dag 15 og videre under hele behandlingsforløpet. Også etter eventuell stamcelletransplantasjon gis imatinib i ett år. Under behandlingen monitoreres tilstedeværelsen av bcr-abl-translokasjonen regelmessig, og resultatene av denne monitoreringen vil avgjøre om pasienten er kandidat for stamcelletransplantasjon eller ikke. Det må understrekes at indikasjonen for stamcelletransplantasjon er under kontinuerlig revurdering. En ny protokoll for Ph+ALL er på vei, men foreløpig ikke godkjent i Norge.
Anbefalinger:
- Ph+ ALL pasienter bør behandles etter EsPhALL-protokollen («EsPhALL amendement for NOPHO») med imatinib og eventuelt stamcelletransplantasjon. Det kreves eget samtykke fra foreldre. (Evidensgrad B)
ALL-residiv
Ca 15 % av barn behandlet for ALL vil senere få residiv. Siden ALL er den hyppigste barnekreftformen, vil det si at ALL-residiv kommer opp i et antall av 5–10 nye tilfeller i Norge årlig. Prognosen etter residiv av ALL er dårligere enn ved første gangs behandling, med opp mot 50 % sjanse til helbredelse ved sene residiver, dårligere ved tidlige residiver. Hvis residivet opptrer tidlig (mindre enn 6 måneder etter avsluttet primærbehandling – høy risiko), vil pasienten tilbys stamcelletransplantasjon etter oppnådd ny remisjon. Ved sene residiver (standard risiko) er det mest vanlig å behandle med kjemoterapi alene, men avhengig av terapirespons vurdert ved MRD, vil det også her kunne bli aktuelt med stamcelletransplantasjon.
Det har tradisjonelt ikke vært noen ensartet behandling av residiv i Norge eller Norden. Mest brukt har den tyske ALL-REZ BFM 2002-protokollen vært, i noen grad også den engelske ALL UKR3-protokollen. Pasienter med residiv av standard risiko behandles nå etter IntReALL-SR-2010-protokollen som ble åpnet i 2014. Denne protokollen blir prinsippene i den tyske og den engelske tilnærmingen sammenlignet. Norge er tilsluttet denne protokollen. I tillegg er det åpnet en høy risiko del av denne protokollen. Det anbefales å rådføre seg med nasjonal ALL koordinator ved alle nydiagnostiserte residiver.
AML
De Nordiske landene har tradisjonelt sett hatt svært gode resultater i behandling av AML hos barn (Hasle et al., 2012; Lie et al., 2005). AML behandles i henhold til NOPHO-DBH-AML 2012-protokollen. Cytostatikabehandlingen er basert på en kombinasjon av antracykliner, cytarabin og etoposid i induksjonsfasen (første 2 kurer), samt høydose cytarabin alene eller kombinert med etoposid eller anthracyklin i konsolideringskurene. Måling av MRD (minimal restsykdom) ved flowcytometri (evt. ved PCR dersom det ikke er mulig å følge MRD via flow) er kommet inn som stratifiserende faktor i den nye protokollen. Pasientene klassifiseres som standard risiko (SR) eller høy-risiko (HR) basert på behandlingsrespons på de første 2 kurene. Pasienter med > 15 % blaster etter 1. kur samt pasienter med intermediær respons (0,1 til 4,9 % blaster etter kur 2) stratifiseres til HR gruppen. Pasienter med ≥ 5 % blaster etter kur 2 går ut av protokollen og behandles individuelt med ett av flere mulige salvage-regimer. Også pasienter med FLT3 ITD mutasjon (med NPM1 wild type) stratifiseres som HR. SR-gruppen behandles med totalt 5 svært intensive cytostatikakurer. HR-gruppen tilbys stamcelletranplantasjon i første remisjon, vanligvis etter 3. eller 4. kur.
Protokollen inneholdt fra starten av 2 randomiseringer. I første induksjonskur ble anthracyklinene mitoxantron og liposomalt daunorubicin sammenlignet med hverandre. Denne randomiseringen er nå avsluttet og anbefalingen er å bruke MEC (kuren som inneholder Mitoxantron) hos alle pasienter, da denne kuren har gitt signifikant forbedret overlevelse. I andre induksjonskur randomiseres fortsatt mellom kur ADxE (cytarabin, liposomalt daunorubicin, etoposid) og kur FLADx (fludarabin, cytarabin, liposomalt daunorubicin). Pga verdensomspennende mangel på Daunoxome, kan Daunoxome erstattes med Daunorubicin (mens Daunoxome ikke er tilgjengelig). Den nordiske protokollen inneholder høye kumulative antracyklindoser, og regelmessige ekkokardiografiske kontroller som spesifisert i protokollen er svært viktig (Lie et al., 2005) (Evidensgrad C).
En undergruppe av AML kjennetegnes ved t(15;17) (PML-RARA) i de leukemiske blastene (akutt promyelocyttleukemi – APL, FAB-klassse M3). Disse pasientene har ofte en uttalt koagulopati på diagnosetidspunktet, som ved DIC, og de krever umiddelbar behandling med «all-trans-retinoic acid» – ATRA, som raskt vil bedre blødningstilstanden. Pasienter med APL har etter et vedtak i NOPHO blitt behandlet etter protokoll «ICC APL Study 01», men en ny protokoll for SR APL er under utarbeidelse (ICC APL study 02). Den nye protokollen anbefales brukt allerede nå (se anbefaling på https://www.nopho.net/ under protokoller/APL). Denne nye protokollen kjennetegnes ved bruk av arsen-trioxyd (ATO) i tillegg til høydosert vitamin A (all-trans-retinoic acid, ATRA, = Vesanoid®) og inneholder ikke lenger vanlige cytostatikakurer. PCR-baserte MRD-undersøkelser underveis i behandlingen er svært viktige, da det finnes en effektiv residivbehandling som er skissert i samme protokollen. Akutt promylocyttleukemi er så ekstremt sjeldent i Norge (< 1 tilfelle per år hos barn) at man bør ta kontakt med nasjonal AML koordinator/NOPHO AML gruppen dersom et tilfelle er diagnostisert eller mistenkt.
Anbefalinger:
- Pasienter med AML bør behandles etter den til enhver tid gjeldende NOPHO-protokollen, for tiden NOPHO-DBH-AML 2012 protokollen. Det kreves eget samtykke fra foreldre.
- Pasienter i høyrisikogruppen (inkl. pasienter med FLT3 ITD og samtidig NPM1 wild type) bør tilbys allogen stamcelletransplantasjon i første remisjon.
- Regelmessige ekkokardiografiske kontroller er spesifisert i protokollen som svært viktig.
- Undergruppen av AML (FAB M3, PML-RARA) skal behandles etter internasjonal APL protokoll med med modifikasjoner – ta kontakt med NOPHO-AML gruppen.
Myeloid leukemi ved Downs syndrom
Dette er en egen leukemiform som bare finnes hos barn med Downs syndrom, og da bare i alderen 0–4 år. I blastene påvises alltid GATA-1 mutasjonen. Mange av barna har gjennomgått transitorisk abnorm myelopoiese (TAM) i nyfødtperioden, og av Down-barn som har TAM i nyfødtperioden, vil ca. 25–30 % få AML senere. Diagnosen forutgås ofte av en periode med cytopeni, mest typisk trombocytopeni.
Barn med Down syndrom har nedsatt toleranse for en del cytostatika, men det er dokumentert at de har svært god prognose med en overlevelse opp mot 90 %, selv om de får redusert behandling i forhold til andre barn med AML (Abildgaard et al., 2006; Creutzig et al., 2005; Zeller et al., 2005) (Evidensgrad C). Av den grunn har NOPHO anbefalt å følge den europeiske protokollen «ML-DS2006». Behandlingen i denne protokollen er basert på konvensjonell AML-behandling, men inneholder mindre anthracykliner, og det gis kun 4 kurer.
Anbefalinger:
- Den europeiske ML-DS 2006-protokollen anbefales brukt (Evidensgrad C).
Kronisk Myelogen Leukemi (KML)
KML hos barn er sjelden, men forekommer. Barna behandles etter samme retningslinjer som de voksne med imatinib, men det er foreløpig uavklart om de kan klare seg uten stamcelletransplantasjon på lang sikt. Ved nedsatt respons på imatinib, primært eller sekundært, kan man forsøke annen-generasjons tyrosin kinasehemmer som brukes hos voksne (nilotinib eller dasatinib) (Andolina, Neudorf, & Corey, 2012). Det arbeides med å opprette et internasjonalt register for barn med KML.
Anbefalinger:
- KML hos barn behandles med imatinib som hos voksne.
- Som andrelinjebehandling anbefales nilotinib eller dasatinib.
- Det er foreløpig uavklart hvilken plass stamcelletransplantasjon vil komme til å få i behandlingen i fremtiden.
Juvenil myelomononcyttleukemi (JMML)
En sykdom som rammer fortrinnsvis små barn. Utredningen er detaljert beskrevet i protokollen «EWOG-MDS 2006». Ved mistanke om JMML skal spesialprøver sendes til laboratoriet i Freiburg, Tyskland, rekvisjoner og adresser finnes i protokollen. Barn med JMML kan bare helbredes med hematopoietisk stamcelletranplantasjon. Man prøver å unngå å gi kjemoterapi før transplantasjonen, men noen ganger er dette nødvendig pga svært høye celletall, respirasjonsproblemer eller andre komplikasjoner (B. Gustafsson, Hellebostad, Ifversen, Sander, & Hasle, 2011) (Evidensgrad C). Graft-versus-leukemi-effekten er svært viktig ved JMML, og etter stamcelletransplantasjonen er det derfor nødvendig å redusere og seponere immunsuppresjonen tidlig, helst innen 2–3 måneder. Tilstanden kan forekomme forbigående hos pasienter med «Noonan-syndrom» og løser seg da spontant. Ved JMML anbefales å ta kontakt med nasjonal og/eller nordisk koordinator for EWOG-MDS tidlig.
Anbefalinger:
- JMML pasienter bør behandles etter EWOG-MDS 2006-protokollen.
- Ved JMML er hematopoietisk stamcelletransplantasjon eneste kurative terapi. Man prøver å unngå kjemoterapi før transplantasjonen (Evidensgrad C). Etter stamcelletransplantasjonen bør immunsuppressiv behandling helst seponeres innen 3 måneder.
Non-Hodgkin lymfom
Non-Hodgkin lymfom (NHL) hos barn og ungdom er en heterogen gruppe av maligne tilstander med opphav i det lymfatiske systemet. Sykdomsspekteret varierer fra transformasjon av umodne T- eller B-celleforløpere (lymfoblastiske lymfomer) til neoplasmer av modne, differensierte lymfocytter (perifere T- eller B- celle NHL) (Reiter, 2007). Forekomst av de ulike NHL-subtypene i barne- og ungdomsalderen avviker fra fordelingen hos voksne pasienter.
Hos barn dominerer følgende typer:
- lymfoblastiske lymfomer (ca. 25 % av alle NHL hos barn)
a) med subtyper B- og T-lymfoblastisk lymfom - perifere B-celle NHL (ca. 55 %)
a) med dominans av Burkitt lymfom (ca. 60 % av B-NHL), ellers
b) diffust stor-cellet B-celle lymfom (DLBCL)
c) moden B-celle leukemi (B-ALL)
d) primært mediastinalt B-celle lymfom - perifere T-celle NHL (ca. 15 %)
a) inkludert anaplastisk stor-cellet lymfom (ALCL) (> 90 % av T-NHL).
Alle andre subtyper er svært sjeldne eller opptrer ikke hos barn. Årlig insidens i Norge er ca. 5 pasienter i aldersgruppen 0–14 år (ca. 6 % av alle malignesykdommer hos barn).
Diagnose
Non-Hodgkin lymfom er kjennetegnet av ukontrollert vekst i lymfevev. Hovedsymptom ved NHL er uømme, forstørrede lymfeknuter ved nodal sykdom, ellers en rekke ulike symptomer ved ekstranodale manifestasjoner. Symptomene varierer med lokalisasjon og utbredelse (f.eks. magesmerter ved abdominelt NHL, åndenød ved mediastinalt lymfom). Hyppigste lokalisasjoner er cervikale lymfeknuter, abdomen, mediastinum og ØNH-området, for øvrig bein, beinmarg, CNS, epiduralrom, nyre, testikler, ovarier, hud og bløtdeler. Allmennsymptomer som nattesvette, vekttap, tretthet og feber kan forekomme. Noen pasienter er i nesten upåvirket allmenntilstand, mens andre presenterer seg med livstruende komplikasjoner (f.eks. trakeakompresjon, tverrsnittslesjoner og nyresvikt).
Diagnostikken ved NHL er omfattende og baseres på radiologiske, histopatologiske, immunfenotypiske og i økende grad cytogenetiske og molekylære undersøkelser. Eksakt klassifisering er en forutsetning for å kunne velge korrekt behandlingsprotokoll.
Biopsi av tumormaterialet må være av tilstrekkelig kvalitet og kvantitet, helst en hel lymfeknute dersom dette er mulig uten betydelig risiko. Tumormaterialet sendes til histologi, immunfenotyping (immunhistokjemi, flowcytometri), cytogenetikk (NHL-spesifikke kromosomtranslokasjoner) og molekylærgenetikk (NHL-spesifikke genmutasjoner).
I noen tilfeller vil man kunne stille diagnosen uten operasjon (biopsi), f.eks. ved cytologisk og immunfenotypisk analyse av blod, beinmarg eller pleuravæske.
Obligate undersøkelser i forbindelse med stadieinndelingen inkluderer ultralyd av abdomen, klinisk undersøkelse av testikler (og evt ultralyd), rtg thorax, MR (CT) av alle regioner med forstørrede lymfeknuter, ved CNS symptomer MR caput, MR spinalkanal ved neurologiske utfall, spinalpunksjon, benmargsundersøkelser og ved tidligere hjertesykdom eller symptomer på hjertesykdom ekko cor og EKG. Verdien av PET/CT i NHL-diagnostikken og oppfølgingen er ikke entydig avklart og vil derfor bli evaluert i kommende NHL-studier. Per februar 2019 inngår ikke PET/CT rutinemessig i utredningen av NHL hos barn.
Anbefalinger:
- Obligate undersøkelser i forbindelse med stadieinndelingen inkluderer ultralyd av abdomen, klinisk undersøkelse av testikler (og vt ultralyd), rtg thorax, MR (CT) av alle regioner med forstørrede lymfeknuter, MR caput / spinalkanal ved neurologiske utfall, spinalpunksjon, benmargsundersøkelser, måling av laktat dehydrgenase (LDH) og eventuelt ekko cor og EKG.
Stadieinndeling
Stadium I | En enkel nodal eller ekstranodal tumormanifestasjon uten lokal spredning, bortsett fra mediastinale, abdominale eller CNS/benmargsengasjement. |
Stadium II | En singel ekstranodal manifestasjon med lokal lymfeknutespredning. To eller flere nodale og eller ekstranodale manifestasjoner på samme side av diafragma, med eller uten lokal spredning, bortsett fra mediastinale, epidurale eller utbredte, ikke receserbare abdominale lokalisasjoner, eller benmargsengasjement. |
Stadium III | Lokalisasjoner på begge sider av diafragma. |
Stadium IV | Beinmarg- og/eller CNS-affeksjon (uansett annen lokalisasjon i tillegg). |
Risikostratifisering
B-NHL og ALCL deles inn i risikogrupper. Risikogruppen avgjør behandlingsintensiteten.
B-cellelymfom (iht protokoll B-NHL 2013)
Risikogruppe 1
Makroskopisk fullstendig fjernet tumormanifestasjon
Risikogruppe 2, st I/II
Inkomplett reseksjon. Stadium I + II sykdom
Risikogruppe 2, st III
Inkomplett reseksjon. Stadium III + LDH < 2 x øvre referanseområde
Risikogruppe 3
Inkomplett reseksjon. Stadium III med LD >= 2x øvre referanseområde men < 4x øvre referanseområde, samt stadium IV/B-ALL med LD < 1000 U/l og ingen CNS-sykdom
Risikogruppe 4
Inkomplett reseksjon. Stadium III med LD >=4 x øvre referanseområde, samt stadium IV/B-ALL med LD > = 4x øvrereferanseområde uten CNS-sykdom
Risikogruppe 4, CNS +
Inkomplett reseksjon. Stadium IV/B-ALL inkludert CNS engasjement
Storcellet anaplastisk lymfom (iht protokoll ALCL 99)
Lav-risikogruppe
Sykdom i stadium I som har blitt komplett recesert Standard-risikogruppe.
Inkomplett reseksjon. Ingen hudaffeksjon, ingen mediastinaltumor, ingen affeksjon av lever, milt eller lunge.
Høy-risikogruppe
Inkluderer pasienter med en av følgende:
Hudlesjoner (biopsi), mediastinaltumor, lever-, milt- eller lungeaffeksjon
Behandling
Primærutredningen og ansvaret for behandling av Non-Hodgkin lymfom hos barn og ungdom tilligger de fire barneonkologiske sentraene ved universitetsykehusene. Alle de nordiske land følger samme behandlingsprotokoller i samråd med NOPHO (Nordic Society of Paediatric Haematology and Oncology).
Behandlingsstrategi: lymfoblastiske lymfomer behandles i hht strategien for akutt lymfatisk leukemi (ALL) med tilstrebet kontinuerlig cytostatika kombinasjonsbehandling over en lang periode (18–24 mnd). Ikke-lymfoblastiske lymfomer behandles med kortere, intensive kombinasjonskurer med høy doseintensitet. Ti års overlevelse for hele gruppen av Non-Hodgkin lymfom hos barn er 87–93 % (Nasjonalt kvalitetsregister for barnekreft, 2018) .
Etter internasjonal konsensus deles lymfombehandlingen hos barn inn i 3 terapeutiske hovedgrupper:
- Lymfoblastisk lymfom: I Norge behandles både T- og B-lymfoblastisk lymfom etter den europeiske behandlingsprotokollen LBL 2018. Protokollen er basert på den tidligere behandlingsprotokollen Euro-LB 02. Protokollen er per våren 2019 åpen for inklusjon av norske pasienter. Protokollen inkluderer induksjons-, konsoliderings- og vedlikeholdsbehandling med systemisk og intratekal cytostatika kombinasjonsbehandling som inkluderer vinkristin, prednisolon, deksametason, methotrexat, daunorubicin, doksorubicin, asparaginase, cyklofosfamid, cytarabin, 6-mercaptopurin og 6-thioguanin. Behandlingsintensiteten tilpasses sykdomsutbredelsen (stadium). «Event-free survival» for hele gruppen er ca. 80–90 %.
- Perifere B-NHL: Norge deltar i studieprotokollen B-NHL 2013, som har vært åpen i Norge siden 2018. I protokollen inngår 2 randomiseringer (Norge deltar per februar 2020 kun i en av randomiseringene), og bruk av Rituximab og undersøkelse av immunrekonstitusjon etter Rituximab er en av hovedmålene med studien. Det er fire ulike risikogrupper (RI-RIV) og per februar 2019 får alle pasientene Riuximab som en del av behandlingen. Behandlingen for øvrig inkluderer (i ulik grad avhengig av risikogruppe) vinkristin, vindesin, deksametason, prednisolon, methotrexat, doksorubicin, etoposid, cyklofosfamid, ifosfamid og cytarabin. CNS-positive pasienter får intensivert intrathekal behandling. Stråling inngår ikke i behandlingen. «Event-free survival» for hele gruppen er ca. 85–90 %.
Rituximab er et monoklonalt antistoff rettet mot overflateantigenet CD20 på lymfatiske B‑celler og lymfomceller. To prospektive kliniske studier har de siste årene vist at infusjon av Rituximab har antitumoraktivitet mot modne B- cellelymfomer hos barn – både dersom gitt som eneste medikament og additivt i kombinasjon med standard kjemoterapibehandling (Goldman et al., 2013; Meinhardt et al., 2010). I aktuell behandlingsprotokoll anser man at Rituximab har en viktig rolle i behandlingen av avansert modent B-cellelymfom hos barn.
- Anaplastisk stor-cellet lymfom (ALCL): I Norge behandles ALCL hos barn etter den internasjonale protokollen ALCL99 (pr. feb. 2020). Standardarmen i protokollen (ingen randomisering) brukes nå som «best available treatment». Kombinert intratekal og systemisk cytostatikabehandling som inkluderer deksametason, cyklofosfamid, cytarabin, ifosfamid, methotrexat, etoposide og doksorubicin, ingen randomisering. Behandlingsintensiteten tilpasses sykdomsutbredelsen (stadium). Stråling inngår ikke. «Event-free survival» for hele gruppen er ca. 70 %. En ny europeisk behandlingsprotokoll for ALCL er i avsluttende planleggingsfase, det er usikkert om Norge vil kunne delta aktivt i denne.
Anbefalinger:
- Pasienter med lymfom bør behandles etter den til enhver tid gjeldende behandlingsprotokoll anbefalt av NOPHO-lymfomgruppen for hver av de terapeutiske lymfom- hovedgrupper. Det kreves samtykke fra foreldrene for inklusjon i de aktuelle protokollstudiene og evt i NOPHO registeret.
- For svært sjeldne lymfomgrupper og ved residiv av NHL må behandlingen individualiseres, etter nasjonal og evt internasjonal diskusjon.
Behandling ved residiv
Tidsperioden med størst risiko for tilbakefall varierer med NHL-subtype og er for B-NHL ca. 6–9 mnd fra diagnosetidspunktet, for ALCL ca. 15 mnd. T- lymfoblastisk lymfom (T-LBL) kan residivere svært tidlig under pågående eller rett etter avsluttet behandling, mens residiver av B-LBL kan også opptre etter noen år.
Residiver ved lymfoblastisk lymfom blir ofte behandlet etter protokoller for ALL- residiv, og strategien inkluder allogen stamcelletransplantasjon. Likevel er langtidsprognosen per i dag svært dårlig (toksisitet, progress). Det samme gjelder for residiv ved moden B-NHL (som Burkitt), til tross for behandling med høyintensive cytostatikakombinasjoner og allogen (eller autolog) transplantasjon. Ved moden B-NHL residiv er tidspunktet for transplantasjon kritisk (dvs straks etter oppnådd remisjon), type transplantasjon (allogen vs autolog) er sannsynligvis av mindre betydning for overlevelsen (Minard-Colin et al., 2015). For CNS-tilbakefall av B-NHL er det rapporter som tyder på at Rituximab trygt og effektivt kan gis intraventrikulært/intrathekalt (Ceppi et al., 2016). Prognosen ved ALCL residiv er klart bedre enn ved andre NHL-subtyper, og kan ofte kureres ved monoterapi med Vinblastin, men i noen tilfeller er det nødvendig med allogen stamcelletransplantasjon og i motsetning til andre B-NHL residiv er det ikke like viktig med fullstendig sykdomsremisjon før transplantasjon. ALCL residiv behandles etter ALCL relapse 04 (med amendment 2013). Det er kommet en del nyere medikamenter som har vært brukt i ulike NHL residiv sammenhenger (ALK hemmere, Brentuximab Vedotin, Blinatumomab, CAR-T m.fl). Disse vil bli vurdert i hvert enkelt tilfelle av residiv og det foreligger ingen standard indikasjon for disse.
Kontrollopplegg for Non-Hodgkin lymfom
Avhengig av NHL-subtype. Hver protokoll inneholder som oftest detaljert beskrivelse av kontrollenes hyppighet og omfang av diagnostikk. Orienterende og veiledende gjelder:
Billeddiagnostikk av affisert område (helst ultralyd, men avhengig av lokalisasjon evt. MR, CT, rtg):
- Lymfoblastisk lymfom: Residiverer tidlig, derfor rtg thorax hver 3. mnd. i vedlikeholdsfasen; i 1. året etter behandling billeddiagnostikk hver 3. mnd, i 2. året hver 6. mnd, deretter kun ved mistanke om tilbakefall. BM og CNS-diagnostikk kun ved mistanke om residiv.
- B-NHL: hver 3–6. mnd etter avsluttet terapi første 18 mnd (avhengig av protokoll); deretter hver 6. mnd frem til (2-)3 år etter behandlingsslutt. Deretter kun ved mistanke om tilbakefall. BM og CNS-diagnostikk også kun ved mistanke om residiv.
- ALCL: hver 3. mnd etter avsluttet terapi første året, hver 4. mnd andre året, hver 6. mnd i totalt 5 år.
Klinisk undersøkelse og blodprøver:
- Hver 1–3 mnd. første år etter avsluttet terapi, deretter hver 3.–6. mnd, avhengig av protokoll.
Ekkokardiografi/nyrefunksjon/gonadefunksjon:
- Avhengig av behandling.
Hodgkin lymfom
Hodgkins lymfom (HL) er et malignt lymfom utgående fra B-lymfocytter. HL består av to entiteter; klassisk HL (med undergruppene lymfocyttrik type, nodulær sklerose, bladet cellularitet og lymfocyttfattig type) og nodulært lymfocyttrikt HL (tidligere paragranulom). Klassisk HL karakteriseres histologisk av et lite antall tumorceller [Hodgkin/Reed-Sternberg (HRS)–celler] i et vev dominert av ikke neoplastiske celler. Ved nodulær lymfocytt rikt HL finnes de såkalte «lymphocyte predominant» (LP)-celler. Det diagnostiseres ca. 140 nye tilfeller av HL pr. år i Norge (Larsen, 2018). En insidenstopp observeres hos unge voksne i aldersgruppen 15–34 år, en annen hos eldre over 50 år. Det er ca. 10 tilfeller hos barn og ungdom under 18 år årlig. Klassisk HL utgjør ca. 95 % av tilfellene og nodulært lymfocyttrikt HL 5 %.
Overlevelsen ved HL har bedret seg betydelig gjennom de siste 50 år. Dette skyldes effektive cytostatikaregimer og bedret bruk av strålebehandling. Siden 1998 har Norge deltatt i studier i regi av Deutsche Arbeitsgemeinschaft für Leukemie (DAL), senere Gesellschaft für Pädiatrische Onkologie und Hämatologie (GPOH). Fra 2008 er dette videreutviklet til et europeisk samarbeid, EURONET, der Norge deltar. Med disse protokollene er prognosen for barn og ungdom med HL generelt svært god.
Diagnose
Klassiske symptomer ved HL er glandelsvulst og vekttap/fravær av adekvat vektøking for alderen, feber, patologisk nattesvette (såkalte B-symptomer) eller andre allmennsymptomer, f.eks hudkløe. Ved undersøkelse legges spesielt vekt på patologisk glandelsvulst og hepato-splenomegali. Blodprøver er ikke spesifikke for HL, men viser ofte bl.a. høy SR, leukocytose med granulocytose, lymfopeni, anemi, trombocytose, høy CRP og gjerne lav albumin. Diagnosen stilles ved en skjærebiopsi av affisert lymfeknute eller tilsvarende. Det er viktig å være klar over at finnålsaspirasjons-cytologi alene kan være falsk negativ ved mange lymfomer, spesielt ved HL, og at cytologi aldri alene er godt nok for å diagnostisere HL. For histopatologisk undersøkelse, se Nasjonalt handlingsprogram med retningslinjer for diagnostikk, behandling og oppfølging av maligne lymfomer (Helsedirektoratet, 2019).
Utbredelsesdiagnostikk ved HL inkluderer minimum en full anamnese med blant annet fokus på B-symptomer, klinisk undersøkelse, bredt anlagte blodprøver (inkludert senkningsreaksjon (SR)), ØNH-undersøkelse (kan utelates), røntgen thorax (kan utelates), ultralydundersøkelser av abdomen, MR av hals, thorax, abdomen og bekken samt CT av thorax. PET/CT er en standard modalitet for diagnostikk og måling av behandlingseffekt. PET-CT vurdert av trenede nuklærmedisinere kan erstatte benmargsbiopsi, som ellers skal tas. (for detaljer se Nasjonalt handlingsprogram med retningslinjer for diagnostikk, behandling og oppfølging av maligne lymfomer (Helsedirektoratet, 2019).
Ann Arbor klassifikasjonen brukes for stadieinndeling av HL og danner basis for behandlingsgrupper (se nedenfor).
Stadium I | Sykdom i en lymfeknuteregion (milt, thymus og Waldeyers ring regnes som nodal affeksjon). |
Stadium II | Sykdom i to eller flere lymfeknuteregioner på samme side av diafragma eller en eller flere lymfeknuteregioner på samme side av diafragma med innvekst i ekstralymfatisk organ/vev (IIE). |
Stadium III | Sykdom i lymfeknuteregioner på begge sider av diafragma eller en eller flere lymfeknuteregioner på begge sider av diafragma med innvekst i ekstralymfatisk organ/vev (IIIE). |
Stadium IV | Diffus eller disseminert sykdom i et eller flere ekstralymfatiske organ/vev med eller uten affeksjon av lymfeknuter. |
Tilleggsopplysninger til Ann Arbor
- Ekstranodal vekst: Markeres med suffikset E. Lokalisert affeksjon av vev/organ i tilslutning til affiserte lymfeknuter slik at direkte innvekst vekst kan antas.
- Bulky sykdom: Markeres med suffikset X (definisjonen varierer etter protokoll).
- B-symptomer: Inndeling i undergruppene A eller B avhengig av om pasienten ikke har eller har ett eller flere B symptomer:
- Uforklarlig vekttap på mer enn 10 % siste 6 måneder
- Uforklarlig persisterende eller residiverende feber med temperatur over 38 °C siste måned
- Gjentatt kraftig nattesvette siste måned.
Risikostratifisering
I den pågående Euronet –PHL-C2 studien defineres 3 terapinivåer (treatment level, TL), basert i hovedsak på stadieinndeling etter Ann Arbor klassifikasjonen. Det skilles mellom 3 nivåer:
TL-1 | Stadium IA/IB/IIA med SR < 30 mm/h og fravær av bulky sykdom her definert som største lesjon over 200 ml. |
TL-2 | Stadium IA/IB/IIA med SR >30 mm/h og/eller bulky sykdom over 200 ml, stadium IAE/IBE/IIAE/IIB/IIIA |
TL-3 | Stadium IIBE/IIIAE/IIIB/IV |
Anbefalinger diagnostikk:
- Diagnosen stilles ved en skjærebiopsi av affisert lymfeknute eller tilsvarende.
- Utbredelsesdiagnostikk som anført.
- PET/CT er vesentlig før, under og etter behandling.
- Benmargsbiopsi kan utelates dersom PET-CT evaluerer benmargsaffeksjon.
Hodgkins lymfom – behandling
Ansvaret for behandling av HL tilligger universitetsykehusene. Behandling ved nodulært lymfocyttrikt HL og klassisk HL er noe forskjellig, og de omtales derfor hver for seg.
Behandling ved nodulært lymfocyttrikt Hodgkins lymfom
Denne subgruppen av HL presenterer seg regelmessig i stadium I eller II. Ikke sjelden er all synlig tumor fjernet ved kirurgi for biopsitaking. Data taler for at slike pasienter kan observeres uten videre behandling (Mauz-Korholz et al., 2007) (evidensgrad C). De fleste residiver etter kirurgi alene vil være lokaliserte og ikke føre til et høyere stadium eller mer komplisert behandling for pasientene enn om de fikk onkologisk tilleggsbehandling første gang. En europeisk studie, Euronet-PHL-LP1, startet i 2010 for blant annet prospektivt å evaluere denne tilnærmingen ved kirurgisk resesert sykdom. Norge deltar ikke i denne studien per i dag. For pasienter med restsykdom etter kirurgi, residiv etter komplett kirurgisk reseksjon eller utbredt sykdom blir behandlingen lik den som gis for tilsvarende stadier av klassisk HL.
Sene residiver etter kjemoterapi og strålebehandling forekommer ikke sjelden, og de har ofte et indolent preg. Standard terapi baseres på samme prinsipper som ved klassisk HL, selv om det mer usikkert om man kan oppnå varig kurativ effekt. Nytten av HMAS er heller ikke godt dokumentert. Regimer som IEP, ABVD, LVPP eller MIME kan forsøkes. Det kan være aktuelt å gi disse regimene kombinert med rituximab siden lymfomet er CD20 positivt. Rituximab monoterapi kan forsøkes idet det er dokumentert responsrater hos voksne (Schulz et al., 2008). Stråleterapi mot problemområder forventes å ha god effekt.
Anbefalinger:
- Kirurgi anbefales når tumorreseksjon er mulig (Evidensgrad C).
- Ved residiver/ikke resesert sykdom gis tilsvarende kjemoterapi som ved klassisk Hodgkins lymfom (se nedenfor).
- Ved sene residiver kan det være aktuelt å prøve rituximab monoterapi eller som kombinasjonsbehandling (Evidensgrad C).
Behandling ved klassisk Hodgkins lymfom
For barn og ungdom er det viktig at den kumulative dose av hvert enkelt cytostatikum er minst mulig, og at strålefeltene og -dosene reduseres i størst mulig grad. DAL/GPOH gruppen har gjennomført suksessive protokoller der behandlingen er redusert gradvis men tilpasset sykdomsutbredelsen. Kurasjonsratene er opprettholdt. Den avsluttede GPOH-HD 95 protokollen (Dorffel et al., 2013) er nylig publisert med lang oppfølgingtid og viser hva som kan oppnås ved disse protokollene.
I Norge er barn og unge med Hodgkins lymfom siden 2008 behandlet i en europeisk protokoll (EuroNet-PHL-C1). Studien etablerte 2 initiale OEPA kurer (vinkristin, etoposid, prednisolon og doksorubicin) som behandling til alle. I TG 2 og 3 (mer utbredt sykdom) viste en randomisert undersøkelse at videre 2–4 kurer COPDAC-28 (cyclofosfamid, vinkristin, prednisolon og dakarbacin) anses som standard (Mauz-Korholz et al., 2010) (Evidensgrad A). PET-CT ble innført som evaluering før og etter to OEPA kurer som instrument for å avgjøre hvilke pasienter som skal ha supplerende stråleterapi. Ikke publiserte data viser at pasienter med negativ PET-CT etter 2 kurer OEPA (interim PET-CT) som ikke får strålebehandling stort seg har samme helbredelsesrater som pasienter med positiv interim-PET-CT og som gis supplerende strålebehandling (Gallamini et al., 2007) (Evidensgrad C).
Studien med endringer (Pasienter med stadium IA/B eller IIA med SR>30 og/eller største tumorvolum over 200 ml flyttes til TL 2 og COPDAC-28 for alle i TL 2 og 3) danner standardarmene i den nye Euronet-PHL-C2 studien.
Euronet-PHL-C2 studien startet januar 2016. Studien har som mål å redusere bruken av strålebehandling ytterligere ved bruk av mer intensiv kjemoterapi. Det stratifiseres i terapinivåer TL-1, 2 og 3 (se ovenfor). I TL-1 gis OEPA kurer og strålebehandling dersom interim PET-CT er positiv. Om interim PET-CT er negativ, gis en tredje kur som COPDAC-28. I TL-2 og –3 randomiseres til en standard arm der videre kjemoterapi er 2–4 COPDAC-28 eller en eksperimentell arm bestående av 2–4 kurer med tillegg av doxorubicin og etoposid (DECOPDAC-21). For pasienter som er interim PET-CT negative gis ingen strålebehandling. For pasienter som er interim PET-CT positive i standard armen gis strålebehandling etter de samme prinsipper som før. For pasienter med positiv interim PET-CT i den eksperimentelle armen gis kun strålebehandling mot områder med PET opptak etter endt kjemoterapi.
Opptak klassifisert som Deauville score 1–3 regnes som negativt både ved interim-PET–CT og PET-CT etter endt behandling.
Behandling ved residiv
Helbredelse er fortsatt mulig ved første residiv, og i noen tilfeller også ved senere residiver. Prognosen er bedre desto lengre tid etter primærbehandlingen residivet oppstår. Euronet-PHL-C1 studien har en egen residivbehandling som vil egne seg for mange pasienter etter tidligere behandling iht GPOH-HD-95 eller Euronet- PHL-C1/2. Behandlingen innebærer konvensjonelle kurer konsolidert med strålebehandling for pasienter med sene residiver etter den minst omfattende primærbehandlingen, helst også uten strålebehandling primært. For de aller fleste andre vil konvensjonelle kurer etterfulgt av HMAS og eventuelt også strålebehandling være aktuelt ved første residiv (Schellong et al., 2005) (Evidensgrad C). HMAS vil også være aktuelt hos pasienter ved 2. eller senere residiv som ikke har fått HMAS tidligere, og som fortsatt har kjemosensitiv sykdom. Brentuximab vedotin er aktuelt som del av induksjonsbehandlingen og som vedlikeholdsbehandling for en del pasienter med risikofaktorer ved residiv (Moskowitz et al., 2018).
Ved residiv etter HMAS vil behandlingsmålet være å oppnå en ny remisjon for mulig å kunne gjennomgå en allogen stamcelletransplantasjon med familiegiver eller ubeslektet giver. Flere konvensjonelle kurer kan velges, i nyere tid også brentuximab vedotin eller immunterapi med check point inhbiotrer (nivolumab og pembrolizumab (Ansell et al., 2015). Dersom kurativt siktemål ikke lenger er mulig, vil stoffer som gemcitabin, vinorelbin, steroider og trosfosfamid være aktuelle med den hensikt å gi responser, palliasjon og evt. Livsforlengelse, evnt kurer som CEKP eller BOP. Lokal strålebehandling kan være en god palliativ behandling ved kjemoresistent sykdom.
Anbefalinger:
- Barn og ungdom med klassisk Hodgkins lymfom bør tilbys inklusjon i EuroNet-PHL-C2 protokollen, alternativt følge EuroNet-PHL-C1 som terapianbefaling.
- Høydosebehandling med autolog stamcellestøtte kan være aktuell behandling ved residiver.
- Residivbehandlingen har endret seg mye de siste år etter at brentuximab vedotin, nivolumab og pembrolizumab er blitt godkjent for bruk.
- I utvalgte tilfeller av residiver etter høydosebehandling og som på ny har vist god respons på kjemoterapi, kan allogen stamcelletransplantasjon være et alternativ.
Kontrollopplegg for Hodgkins lymfom
Euronet-PHL-C1 studien har spesifikke planer for oppfølging, som i hovedsak gjenspeiler hva en kan anse som rutine for disse pasientene. Kontroller foretas hver 3 mnd i starten, deretter hver 4 mnd, etter hvert hver 6–12 mnd. Klinisk undersøkelse og blodprøver gjøres ved alle kontroller. Billedundersøkelser gjøres ved fastlagte kontrolltidspunkt. Funksjonsundersøkelser av thyreoidea, gonader, hjerte eller lunger samt regelmessig mammografi gjøres etter vurdering av den behandling pasienten har fått, spesielt mht strålebehandling mot de respektive organer. Se Nasjonalt handlingsprogram med retningslinjer for diagnostikk, behandling og oppfølging av maligne lymfomer (Helsedirektoratet, 2019).
Seneffekter, spesielt etter strålebehandling, kan komme sent og pasienter som har fått store strålefelt mot mediastinum bør kontrolleres livslangt. Kontrollene kan etter 5–10. år overføres til lokalsykehus etter retningslinjer gitt fra spesialavdeling, og etter hvert til fastlege. Det er viktig med kompetanse og interesse for denne pasientgruppen ift å påvise residiv tidlig og å kjenne til langtidsbivirkningene etter behandling hos denne relativt unge pasientgruppen (Helsedirektoratet, 2019).
Histiocytoser
Langerhanscellehistiocytose (LCH)
Histiocytoser er en gruppe sykdommer som skyldes unormal proliferasjon av celler tilhørende monocytt-/makrofagsystemet. Ved langerhanscellehistiocytose (LCH) er det langerhanscellene som er den patologiske cellepopulasjonen. Langerhansceller er benmargsderiverte celler som naturlig finnes i hud og lymfeknuter. De prolifererende cellene er vist å være av klonal opprinnelse, men LCH regnes ikke som en kreftsykdom. Den har dog i ICD-10 fått et C-nummer. Nyere molekylær-genetisk forskning fra de siste årene viser i tillegg at en betydelig andel av de patologiske langerhanscellene har mutasjoner i BRAF eller MAP2K genene. Dette har betydning for mulig behandling.
Inndeling
LCH deles inn etter sykdommens utbredelse i enkelt- eller multisystemtype på følgende måte:
- Enkeltsystem-LCH
- Enkeltsystem – én lokalisasjon (single system – single site)
- Enkeltsystem – flere lokalisasjoner (single system – multiple site)
- Enkeltsystem – spesiell lokalisasjon (single system – special site)
- Multisystem-LCH
- Multisystem uten affeksjon av risikoorganer
- Multisystem med affeksjon av risikoorganer (lever, milt, benmarg)
Epidemiologi
Sykdommen er svært sjelden med en anslått insidens på 8–9 tilfeller pr. million barn pr. år, tilsvarende for voksne (Abla, Egeler, & Weitzman, 2010). Det er antatt at dette er et minimumstall, siden enkeltstående lesjoner med spontan tilbakegang ikke alltid blir diagnostisert eller rapportert.
Organmanifestasjoner
- Hud – utslett som ligner seborroisk eksem er vanlig, særlig hos de minste barna. Utslettet er ofte lokalisert i hodebunnen, bak ørene, i øregangene, i hudfoldene i axiller og lysker, nederst på ryggen, i flankene og nederst på abdomen.
- Skjelett – smertefulle lesjoner i skjelettet, ofte i kalvariet eller de lange rørknoklene.
- Lymfeknuter – infiltrasjon av en eller flere lymfeknuter med patologiske langerhansceller. Forstørrede lymfeknuter kan enten opptre som isolert fenomen eller som ledd i mer generalisert sykdom.
- Lever, milt – ofte forstørret som ledd i multisystemsykdom.
- Benmarg – infiltrasjon av benmargen med patologiske langerhansceller. Benmargsaffeksjon som risikokriterium er definert som nedsatt produksjon av hematopoietiske celler (anemi, leukopeni, trombocytopeni).
- Lunger – alene eller som ledd i multisystemsykdom. Lungeaffeksjon er særlig vanlig hos voksne røykere
- Tarm – tarmaffeksjon er sjelden, men sees iblant.
- CNS
- Diabetes insipidus (DI) er svært vanlig ved LCH. DI kan være første manifestasjon, ledd i multisystemsykdom eller sees ved enkeltsystem skjelettsykdom. DI er særlig hyppig som komplikasjon ved skjelett-LCH lokalisert i skallebasis eller ansiktsskjelett.
- Nevrodegenerativ LCH – dette er en tilstand som rammer noen LCH-pasienter. Det er uklart hva det egentlig representerer, ikke nødvendigvis aktiv LCH
Symptomer
Symptomene varierer med sykdommens lokalisasjon og utbredelse.
- Enkeltsystem-LCH:
Det er først og fremst lokalisasjonen som avgjør symptomene: utslett, smertefull kul/hevelse, hovne lymfeknuter etc., se foregående avsnitt - Multisystem-LCH
Her er mer enn ett organsystem affisert, med eller uten allmennsymptomer, med eller uten affeksjon av risikoorganer
Risikoorganer:- Lever
- Milt
- Benmarg (definert som cytopeni i minst 2 linjer)
Tidligere var også lunger definert som risikoorgan, men dette er i den nyeste protokollen tatt ut som risikoorgan.
Karakteristisk for sykdommen er tendensen til spontane remisjoner og eksaserbasjoner.
Diagnose
Radiologisk utredning ved diagnose er røntgen totalskjelett, røntgen thorax og ultralyd abdomen. Ytterligere undersøkelser som CT av lungene og CNS utredning kun på indikasjon.
Diagnosen stilles ved biopsi av en lesjon. De patologiske cellene ved LCH skiller seg fra normale langerhansceller ved at de på overflaten har markøren CD1a, og at de inneholder Birbeck-granula som er synlige ved elektronmikroskopi. For en sikker diagnose kreves påvisning av CD1a og/eller CD207 (Langerin) positive celler. Elektronmikroskopisk påvisning av Birbeck-granula er ikke obligatorisk.
Ved DI som første manifestasjon av LCH vil vi ofte finne en fortykket hypofysestilk ved MR-undersøkelse. Før evt. spesifikk LCH-behandling kreves biopsiverifikasjon.
Behandling
Behandlingen har tradisjonelt vært kontroversiell pga. sykdommens naturlige svært svingende forløp. Det har vært to prinsipielt forskjellige tilnærminger til behandlingen, dels et behandlingsprinsipp som innebærer så lite behandling som mulig pga. den store tendensen til spontane remisjoner, dels et prinsipp om tidlig og intensiv behandling med kjemoterapi for at sykdommen skal falle til ro raskt, for å redusere permanente sekveler og redusere risikoen for reaktivering av sykdommen. Gjennom Histiocyte Society (stiftet i 1985, en internasjonal organisasjon for forskning og behandling av histiocytoser) har man etter hvert lyktes i å komme frem til felles behandlingsretningslinjer basert på resultater fra tidligere studier. Siden 2015 har vi behandlet pasienter i henhold til LCH IV protokollen fra Histiocyte Society.
- Enkeltsystemsykdom
Behovet for aktiv behandling varierer avhengig av hvilket organ som er affisert, og hvilken eller hvilke lokalisasjoner- Hud: Lokal steroidbehandling, evt. peroral etoposid (bare aktuelt til voksne). Hud-LCH kan være ledd i multisystemsykdom, slik at pasienten må utredes med henblikk på andre lokalisasjoner. Evt. kan det skje videreutvikling til eller reaktivering i form av multisystem-LCH.
- Skjelett: Avhengig av lokalisasjon og om det er en enkelt lesjon eller flere. En enkelt lesjon vil ofte gå tilbake spontant eller etter diagnostisk biopsi, evt. injeksjon av lokale steroider. Visse lokalisasjoner krever aktiv behandling for å avverge senskader (f. eks. lesjon i virvelcorpus med risiko for skade av medulla spinalis, lesjoner i skallebasis eller ansiktsskjelett med økt risiko for CNS-affeksjon). Dette er spesifisert i protokollen. Indometacin og bisfosfonater har også vært prøvd ved skjelett-LCH, og indometacin er et behandlingsalternativ i en randomisert arm i LCH-IV. De enkelte lesjoner følges med røntgen i henhold til den aktuelle protokoll.
- Isolert diabetes insipidus: DI som ledd i LCH er som regel irreversibel. DI behandles med desmopressin. Man må ikke la seg friste til å gi spesifikk LCH-behandling uten biopsiverifikasjon. Siden man som regel vegrer seg for å ta biopsi av en lesjon i hypofysestilken, må man lete etter andre lokalisasjoner for biopsitagning, undersøke spinalvæsken med henblikk på tumormarkører (diff.diagnose: germinalcellesvulst) og CD1a pos. celler. Ved verifisert LCH og en tydelig hypofysestilktumor er det indikasjon for systemisk behandling med vinblastin/steroider etter LCH-IV-protokollen.
- Multisystemsykdom (samme behandling for MS-LCH med eller uten riskorgan affeksjon):
Behandling etter LCH-IV protokollen stratum 1. Behandlingen består av en eller to induksjonsblokker med vinblastin x 6 og steroider per os, etterfulgt av en vedlikeholdsbehandling som inneholder pulser med vinblastin/steroider, randomisert med eller uten 6-merkaptopurin og varighet 12 eller 24 måneder (dvs. 4 randomiseringsarmer). - Multisystemsykdom med forverring eller reaktivering av ikke-risiko organer:
Behandling etter LCH-IV protokollen stratum 2. Behandlingen består av behandling med steroider, cytarabin og vinkristin i 24 uker, deretter randomisering til tablettbehandling med indometacin eller purinethol/metotreksat til totalt 2 års behandling. - Multisystemsykdom med forverring eller reaktivering av risikoorganer:
Det er dette som i størst grad tilsvarer den gamle betegnelsen «Letterer-Siwe disease». Dette er den mest alvorlige formen for LCH, og det er i denne gruppen vi finner pasienter med livstruende sykdom og en relativt høy mortalitet. I LCH-II-protokollen (1996–2000) var mortaliteten 75 % hos pasienter som ikke responderte på behandlingen i løpet av 6–12 uker. Disse pasientene behandles nå i henhold til LCH IV protokollens stratum 3. Behandlingen består av høye doser cytarabin og cladribin, supplert etterhvert med purinethol, metotreksat, vinblastin og steroider. Dersom det ikke er respons på denne behandlingen kan allogen benmargstransplantasjon være et behandlingsalternativ.
BRAF/MEK hemmere
Det er økende evidens for god behandlingsrespons med BRAF hemmere (og også etterhvert MAP2K hemmere) der man finner tilgrunnliggende BRAF/MAP2K mutasjoner i langerhanscellene. Derimot har man sett at pasientene har tendens til reaktivering når man avslutter behandlingen. Disse medikamentene er pr i dag ikke innlemmet i LCH IV protokollen, og det er foreløpig ikke nok kunnskap til å vite hvilke pasienter som bør tilbys behandling, og hvor lenge behandlingen bør vare. I dag blir behandling med BRAF/MAP2K hemmere først og fremst anvendt der man ikke oppnår tilfredsstillende behandling med konvensjonell behandling.
Radiologisk oppfølging
Ved CNS risikolesjoner hver 3. måned første år, hver 6 måned 2.–5. År. Ved skjelettlesjoner røntgen hver 3. måned til tilbakegang. Ytterligere oppfølging kun på indikasjon
Prognose
For enkeltsystem-LCH og multisystem-LCH uten affeksjon av risikoorganer er prognosen vedrørende overlevelse svært god (100 %). Det er dog en stor risiko for en eller flere reaktiveringer av sykdommen og for permanente sekveler, først og fremst ortopediske, diabetes insipidus og evt. panhypopituitarisme. Det er også de senere årene blitt klart at enkelte pasienter kan få en nevrodegenerativ tilstand forårsaket av LCH. Denne tilstanden er lite påvirkelig av terapi. I LCH IV protokollen anbefales nå at denne gruppen pasienter med kliniske symptomer på nevrodegenerasjon behandles med enten cytarabin eller immunglobuliner.
Hemofagocytisk lymfohistiocytose (HLH)
Hemofagocytisk lymfohistiocytose (HLH) betegner en hyperinflammatorisk tilstand karakterisert ved langvarig høy feber, cytopenier, hepato-/splenomegali og hemofagocytose i aktiverte, benigne makrofager. HLH kan være primær/genetisk eller sekundær til andre sykdommer, først og fremst infeksjoner, maligne tilstander eller revmatiske lidelser. HLH forekommer både hos barn og voksne. Primær/genetisk HLH opptrer ofte svært tidlig, gjerne i første leveår, mens sekundær HLH vanligvis opptrer senere (Janka, 2007).
Felles for både primær og sekundær HLH er at fagocyterende celler (cytotoxiske T‑lymfocytter (CTL) og NK-celler) som aktiveres etter stimulering av et (infeksiøst) agens, produserer store mengder inflammatoriske cytokiner, noe som normalt vil medføre at det aktuelle patogene agens blir fjernet. Ved HLH har NK- cellene og CTL defekt fagocyterende evne, og cytokinnivået vil derfor holde seg vedvarende høyt og vedlikeholde den inflammatoriske tilstanden.
Diagnosen HLH er ofte vanskelig. Tilstanden omtales her fordi den kan være en vanskelig differensialdiagnose til leukemi hos barn, og fordi den behandles av spesialister i barnehematologi/-onkologi med cytostatika og hematopoietisk stamcelletransplantasjon.
Primær/familiær HLH (FHLH eller FHL) Tilstanden er autosomalt recessivt arvelig med en hyppighet på 1,2:1.000.000 barn pr. år eller ca. 1: 50.000 levende fødte barn i Sverige (Henter, Elinder, Soder, & Ost, 1991). Det er til nå identifisert fire ulike gendefekter som kan gi HLH, mens det hos 50–70 % av pasientene (avhengig av etnisk bakgrunn) ikke kan påvises en kjent gendefekt. Diagnosen er vanskelig, og spesielt tidligere ble diagnosen i en del tilfeller stilt post mortem. Symptomene opptrer ofte i svært ung alder, gjerne tidlig i første leveår, men er også beskrevet høyere opp i barnealder. Selv i arvelige tilfeller er tilstanden ofte utløst av en infeksjon, slik at distinksjonen mellom primær/arvelig og sekundær HLH kan være vanskelig. HLH kan også opptre som ledd i arvelige immunsvikttilstander: Chédiak-Higashis syndrom (CHS), Griscellis syndrom (GS) og X-bundet lymfoproliferativt syndrom (XLP).
Sekundær/reaktiv HLH Tilstanden opptrer sekundært til infeksjoner («virus associated hemophagocytic syndrome» – VAHS (særlig EBV), eller «infection associated hemophagocytic syndrome» – IAHS), maligne sykdommer eller revmatiske lidelser (representerer en litt annen entitet, «macrophage activation syndrome»– MAS). De fleste tilfellene opptrer hos litt større barn eller voksne. Epstein- Barr-virus er antagelig den vanligste utløsende årsaken ved IAHS. Visceral leishmaniasis er en vanskelig og viktig differensialdiagnose, ikke minst fordi infeksjonen er i ferd med å bre seg i Europa (Ready, 2010). Leishmaniainfeksjon kan også være en årsak til sekundær HLH. Hemofagocytose kan også sees som et mer uspesifikt reaktivt fenomen, f. eks. hos intensivpasienter med multiorgansvikt (Strauss et al., 2004).
Diagnostiske kriterier (enten 1. eller 2. må være oppfylt) (Henter et al., 2007):
- Molekylær diagnose forenlig med HLH
- Diagnostiske kriterier for HLH til stede (minst 5 av 8):
- Opprinnelige kriterier:
- Feber
- Splenomegali
- Cytopeni i minst 2 cellelinjer i perifert blod
(Hb < 9 g/dL, tr.c. < 100 x 109/L, nøytrofile < 1,0 x 109/L) - Hypertriglyceridemi og/eller hypofibrinogenemi
(fastende triglycerider ≥ 3,0 mmol/L, fibrinogen ≤ 1,5 g/L) - Hemofagocytose i benmarg, milt eller lymfeknute
(ofte ikke til stede tidlig i sykdomsforløpet). Ingen tegn til malignitet
- Nye kriterier:
- Lav eller opphevet NK-celleaktivitet
- Ferritin ≥ 500 μg/L
- Løselig CD25 (IL-2-receptor) ≥ 2400 U/ml
- Opprinnelige kriterier:
Hemofagocytose som har gitt sykdommen dens navn, er ofte ikke til stede tidlig i forløpet, og det kan også sees ved andre tilstander. Det er derfor viktig å ha en høy grad av mistenksomhet overfor HLH hos alvorlig syke, septiske sped- og småbarn som har cytopenier, hepato-/splenomegali og manglende effekt av antibiotikabehandling. Mange har CNS-affeksjon med forhøyet celletall i spinalvæsken. Det er avgjørende at de riktige prøvene blir tatt, først og fremst ferritin, triglycerider og koagulasjonsstatus inkludert fibrinogen. Undersøkelse av interferon og interleukiner kan være til hjelp for å stille diagnosen, (IL-2 reseptor analyseres bl.a på Hormonlaboratoriet på Aker (OUS) (0.5 ml frossent serum)). Det må også gjøres benmargsundersøkelse for å utelukke leukemi og eventuelt påvise hemofagocytose.
Behandling
Eventuell utløsende infeksjon må behandles. Ved EBV infeksjon, kan Rituximab være av nytte. EBV-indusert HLH kan bli svært alvorlig og få dødelig utfall.
Det er utarbeidet en protokoll for behandling av HLH gjennom Histiocyte Society (Henter et al., 2007). Den første protokollen kaltes HLH-94-protokollen, den er senere erstattet med HLH-2004-protokollen der det bare er gjort relativt små endringer.
Behandlingen består av steroider (dexametason) kombinert med etoposid og cyklosporin A i induksjonsfasen over 8 uker. Ved CNS-affeksjon som vedvarer etter 2 ukers behandling, gis intratekal behandling med metotreksat og prednisolon. Ved primær HLH går man etter 8 uker rett over i en vedlikeholdsfase med de samme medikamentene, frem til det kan utføres hematopoietisk stamcelletransplantasjon (SCT) som er eneste kurative behandling. Dersom man ikke oppnår remisjon blir 2. linje behandling forsøkt, f.eks tillegg av antithymocytt-globulin, og individuell tilpasning av behandlingen må gjøres. Hvis man mistenker at det er en sekundær HLH (mildere forløp, høyere alder, ingen familieanamnese eller kjent gendefekt), vil man seponere behandlingen etter 8 uker dersom det er god behandlingsrespons, og følge nøye opp om det kommer et residiv eller ikke. Ved dårlig behandlingsrespons eller tilbakefall må man behandle som primær HLH med SCT, men mange vil vise seg å være helbredet etter de første 8 ukene. Ved mindre alvorlige tilfeller av sekundær HLH kan det være tilstrekkelig med god støttebehandling og immunmodulerende behandling, men terskelen for å behandle etter HLH-2004-protokollen må være lav, siden også den sekundære formen kan ha et raskt progredierende forløp med fatal utgang. HLH-protokollen ble lukket ved årsskiftet 2011–2012, men det anbefales at den brukes fremdeles med en liten modifikasjon i form av utsatt start av cyklosporin til dag 14.
Per februar 2020 pågår det en metodevurdering av Emapalumab (anti-interferon-gamma) til bruk ved refraktær primær HLH. Dette medikamentet ble FDA-godkjent november 2018, og det er en pågående pediatrisk investigational plan («PIP») i Europa. Ved evt behov for medikamentet i en refraktær situasjon før markedsføringstillatelse, kan man henvende seg til produsent for å forhøre seg om muligheten for Compassionate Use, etter retningslinjer i Helseforetaket.
Prognose
Før behandlingen etter HLH-protokollene ble etablert, var den primære formen for HLH alltid fatal, men med behandling som inkluderer SCT, overlever over 50 % av pasientene. Også sekundær HLH kan ha et alvorlig forløp med fatal utgang.
Svulster i sentralnervesystemet
Sist faglig oppdatert: 26.05.2020
Lavgradig gliom
Lavgradige gliomer (LGG) er fellesnavn på svulster utgående fra gliaceller med malignitetsgrad 1 og 2 etter WHO-klassifiseringen (Louis et al., 2007). Hos barn er nesten alle disse astrocytomer. LGG skiller seg fra høymaligne svulster ved at tumorveksten hos de fleste er langsom, og hos noen kan endog veksten stoppe spontant uten behandling. Forløpet kan være fluktuerende med periodevis økning og regresjon av tumorstørrelse, hos andre ses et mer aggressivt forløp og endog metastasering til hjernehinner. Som følge av LGGs natur har pasientene en meget høy overlevelse (OS, Overall Survival), mens progresjonsfri overlevelse (PFS) er mye lavere, dvs at progresjon er vanlig, men lar seg oftest vellykket behandle. Barn under ett år har tendens til mer negativt forløp enn eldre barn. Forskjellige histopatologiske undergrupper av LGG har også forskjellig prognose. Svulstenes lavgradige natur er en meget viktig faktor ved vurdering av behandlingsvalg.
Det er nylig publisert europeiske retningslinjer for diagnose og behandling av LGG hos barn og ungdom (A. K. Gnekow et al., 2019).
Epidemiologi
LGG er de vanligste CNS-svulster hos barn og utgjør ca. 40 % av disse. Pilocytisk astrocytom (PA) er den hyppigst forekommende svulst av alle typer registrert i barnekreftregisteret, ca. 10 % av alle pasienter totalt registrert (Stiller, 2007).
LGG er sterkt assosiert med nevrofibromatose 1 (NF-1), og NF-1-pasienter har særlig overhyppighet av pilocytiske astrocytomer i synsveiene (A.K. Gnekow, Packer, & Kortmann, 2004; Stokland et al., 2010). Ofte er disse multifokale med involvering av hjernestammen og andre strukturer.
Histopatologiske typer og lokalisering
Den helt dominerende typen LGG hos barn er pilocytiske astrocytomer (PA) (WHO grad 1) som utgjør over 80 % av gruppen(Stokland et al., 2010). Diffuse astrocytomer grad 2 (DA) er vanligst hos voksne, men utgjør knapt 10 % hos barn, noe hyppigere hos ungdommer. En variant av PA, pilomyxoid astrocytom (PMA) (WHO grad 2) har et mer aggressivt forløp.
Oligodendrogliomer er svært sjeldne hos barn. Diffuse ponsgliomer inkluderes ikke i LGG selv om enkelte av disse histololgisk er DA (WHO grad 2).
Blandingssvulster av nevronale og gliale celler er sjeldne. De er nesten alltid histologisk lavgradige og omtales oftest sammen med LGG. Den vanligste typen er gangliogliom.
LGG kan oppstå i alle deler av CNS, mest hyppig i cerebellum, synsveiene (n. opticus, chiasma, sentrale synsbaner) og hemisfærene (Stokland et al., 2010). Lokalisasjon varierer med alder. Små barn har overvekt av synsveigliomer mens større barn tenderer mot hemisfærebeliggenhet. Pilocytiske astrocytomer er den dominerende typen i alle aldersgrupper, mens fibrillære er noe hyppigere hos ungdommer enn hos små barn. I synsveier og cerebellum finnes nesten utelukkende PA, mens DA utgjør ca. 10–15 % i andre lokalisasjoner.
Molekylærbiologi
De senere år har en funnet svært viktige forandringer molekylærbiologisk hos barn med LGG (Packer et al., 2017). Det gjelder særlig i signalveien MAPK (RAS/RAF/MEK/ERK) som er viktig for cellenes proliferasjon. Spesifikke endringer i denne signalveien kan gi overaktivitet og dermed økt onkogen stimulering. Pilocytisk astrocytoms biologi regnes i dag å representere en «singel pathway disease». Fusjon av BRAF-genet med annet gen (BRAF-KIAA1549) er vanligst ved PA, mens punktmutasjon av BRAF (BRAF V600E) er sjeldnere og ses ved forskjellige LGG. Det foreligger allerede i dag medikamenter som har LGG med slike mutasjoner som mål (MEK-inhibitorer og BRAF-inhibitorer). Se senere.
Diagnose og utredning
LGG gir symptomer enten pga lokal påvirkning eller pga økt intrakranielt trykk, og bildet vil variere sterkt avhengig av lokalisasjon. Synsfunksjonen er ofte påvirket ved supratentorielle midtlinjesvulster og n. opticus-gliomer. Balanseforstyrrelser ses ved cerebellare svulster, epilepsi ved hemisfærelokalisasjon etc. Økt intrakranielt trykk gir hodepine mest uttalt om morgenen, morgenkvalme og brekninger og noen ganger dobbeltsyn (abducensparese). Hvis symptomene har vart lenge er det tegn på lavgradig svulst, men kort varighet er ikke uvanlig.
MR cerebrum viser tumor. På tross av histopatologisk lavgradighet, kan LGG være disseminert eller multifokal, og derfor skal hele CNS visualiseres. Anbefalte MR-teknikker er som ved andre hjernesvulster hos barn.
Histologisk diagnose er oftest nødvendig, og når radikal reseksjon ikke er mulig, gjøres biopsi. Unntak er synsveigliomer med klassiske MR-funn, særlig hos nevrofibromatose-pasienter, der man godtar radiologisk diagnose uten biopsi, fordi disse svulstene erfaringsmessig alltid er PA. Ved tectum-lokalisasjon vil en også være tilbakeholdende med biopsi.
Molekylærbiologisk utredning er ønskelig ved de fleste LGG både for diagnose og valg av behandling.
Oftalmologisk utredning er svært viktig ved supratentorielle midtlinjegliomer, spesielt i synsveiene. Endokrinologisk kartlegging er vesentlig avhengig av lokalisasjon.
Behandling
Behandling og prognose vil variere med lokalisasjon. Laterale svulster vil ligge bedre til rette for radikal kirurgi, og dermed ha bedre prognose. Midtlinjesvulster vil ikke kunne fjernes radikalt og trenger derfor i mange tilfeller ytterligere behandling, ut fra klare retningslinjer (Perilongo, 2005; Reddy & Packer, 1999).
Kirurgi er primærbehandlingen ved LGG (B. J. Due-Tønnessen et al., 2002). Reseksjonsgrad er den viktigste prognostiske faktor, og radikal reseksjon gir best prognose. Ofte vil radikal kirurgi ikke være mulig pga faren for skade, og da synes ikke partiell reseksjon å gi resultatmessige fordeler vedrørende overlevelse sammenlignet med biopsi (Stokland et al., 2010). Men i en del tilfeller vil partiell reseksjon være hensiktsmessig for å bedre pasientens kliniske tilstand, f.eks. ved hydrocephalus. Tilnærmingen må ses i forhold til mulige skadelige effekter av en partiell reseksjon.
Norge har deltatt i den store internasjonale behandlingsprotokollen SIOP-LGG 2004, som ble avsluttet i 2015. En ny studie (LOGGIC) er nå (2019) på det nærmeste ferdigutformet av SIOP-Europe med randomisering i tre behandlingsarmer. Kombinasjonen Vinkristin/Karboplatin er standardarmen, Vinblastin monoterapi som er mye brukt i dag er andre arm, og den mest eksperimentelle armen vil være MEK-inhibitor (Trametinib). Trametinib gis peroralt. LOGGIC åpner i Norge i 2020. NF1-pasienter inkluderes sannsynligvis ikke i denne fordi denne gruppen skal rekrutteres til en annen studie. Ca. 1/3 av alle LGG-pasienter forventes å trenge ikke-kirurgisk behandling. Det er viktig at klare kriterier oppfylles før slik behandling startes. Dette innebærer at ved diagnose / rett etter primærkirurgi, gis medikamentell behandling bare hvis pasienten har alvorlige nevrologiske symptomer som kan forverres, eller truende synstap. Alle andre pasienter blir observert klinisk og billedmessig. Disse pasientene, som utgjør et flertall, er observasjonsgruppen i studien. Dersom tumor i observasjonstiden vokser signifikant ved MR-undersøkelse, eller det er økende symptomer, vurderes da på ny evt kirurgi. Dersom dette ikke er hensiktsmessig, inkluderes også disse pasientene.
Da også de behandlingstrengende pasientene har god prognose vedrørende overlevelse, innfører LOGGIC nye primære endepunkter, særlig gjelder dette synsfunksjonen. Effektiv behandling som bevarer synet vil være sterkt ønsket. Synsundersøkelse med analyse av funksjonsendring vil derfor være en hovedfaktor i studien.
LGG-pasienter med nevrofibromatose (NF-1) planlegges inkludert i et felles studium med USA. Det ligner LOGGIC, men har bare to armer: vinkristin/karboplatin vs. MEK-inhibitor. Dersom studien ikke lar seg gjennomføre, vil gruppen bli inkludert i LOGGIC.
Strålebehandling kan ha god effekt ved LGG og var tidligere mye brukt. Pga mulige kognitive seineffekter og sekundærkreft, er radioterapi nå ikke en del av primærbehandlingen. Imidlertid kan det være aktuelt når medikamentell behandling ikke er effektiv nok, spesielt ved små svulster og hos eldre barn og ungdom. Strålekniv kan brukes i utvalgte tilfeller. Hos NF-1-pasienter vil en være enda mer tilbakeholdende med strålebehandling pga økt følsomhet for alvorlige strålebivirkninger
En oppnår sjelden komplett remisjon av medikamentell behandling, men varig stabil tumorstørrelse eller noe reduksjon vurderes som tilfredsstillende behandlingseffekt.
Anbefalinger:
- Kirurgi anbefales når radikal tumorreseksjon er mulig, og denne behandlingsmodus skal alltid vurderes først.
- Når radikal kirurgi ikke er mulig, skal biopsi gjøres i de aller fleste tilfellene. Partiell reseksjon synes ikke å gi bedre overlevelsesresultater enn biopsi og er derfor omdiskutert. Dette må tas stilling til i hvert enkelt tilfelle.
- Kirurgi vurderes på ny ved residiv eller progresjon.
- Når klare kriterier er oppfylt (se ovenfor), anbefales å gi nonkirurgisk behandling i henhold til LOGGIC protokollen.
- Pasienter med nevrofibromatose behandles etter egen protokoll.
Ny utprøvende behandling
MEK-inhibitor hemmer MAPK-signalveien «downstream» for BRAF og har vist seg å gi god respons ved LGG i ferske studier. Dette har ført til at trametinib nå skal brukes «up-front» i LOGGIC-studien. Langtidseffekten er foreløpig ikke kjent, men ved behandlingsrefraktær LGG med dårlig prognose bruker mange nå MEK-inhibitor, peroralt en gang daglig. En forventer etter hvert mer kunnskap om virkninger på lengre sikt.
BRAF-inhibitor gir god respons på svulster som har BRAF-punktmutasjon, og er i dag førstevalg når slik mutasjon er påvist. Imidlertid gir den paradoksal effekt i form av økt tumoraggressivitet ved svulster med BRAF-fusjon, og må derfor aldri gis uten bekreftet BRAF V600E punktmutasjon.
Bevacizumab alene eller kombinert med andre midler brukes ved resistent LGG i dag. Den gir god respons hos tidligere sterkt behandlede pasienter, men nesten alle får progresjon etter avsluttet behandling(Hwang et al., 2013). Positiv påvirkning av visus har vært rapportert, men flere studier har ikke kunnet verifisere dette.
Behandling av tilbakefall eller progresjon etter primærbehandling
LGG har et helt annet forløpsmønster enn aggressive kreftformer. Ofte starter tumor ny vekst flere år etter at den har blitt stabilisert av behandling. En kan da gi samme behandling som initialt, eller andrelinjebehandling. I Norge er det for tiden vanligvis vinblastin monoterapi. MEK-inhibitor kan vurderes som anført ovenfor.
Anbefalinger:
- Dersom primærbehandlingen hadde god effekt, kan en gjenta denne.
- Ved tidlig progresjon anbefaler vi annenlinjebehandling. Mest brukt nå er ukentlig vinblastin i ett år (Bouffet et al., 2012).
Organisering av behandlingen
Behandlingen av LGG er regionalisert og ledes av det regionale barnekreftsenteret. Dette gjelder både kirurgi, kjemoterapi og strålebehandling. Deler av kjemoterapien kan gis mer lokalt.
Prognose
Mortaliteten er lav med 5 års overlevelse rundt 95 %, mens progresjonsfri overlevelse (PFS) er atskillig lavere. Komplett reseksjon er den viktigste uavhengige positive prognostiske faktoren, mens alder under ett år ved diagnose og de viktigste grad 2-histopatologiske typene (DA og PMA) er negative faktorer. Ved svulster i synsveiene har NF1-pasienter bedre prognose enn andre (Stokland et al., 2010). Ved synsveigliomer får pasientene ofte senskade i form av betydelig og varig synsreduksjon. DA kan ha en tendens til å utvikle seg til malignt astrocytom etter flere år, men i langt mindre grad enn hos voksne. Dette skjer svært sjelden ved PA, men er beskrevet.
Oppfølging og seineffekter Anbefalinger:
- Klinisk kontroll med MR gjøres hver tredje måned de første to år, deretter hver 6. måned, og deretter årlig.
- Progresjon kan oppstå etter flere år, slik at oppfølging i ca. 10 år anbefales.
- Oftalmologisk oppfølging er essensielt hos pasienter med synspåvirkning.
- Nevropsykologisk testing gjennomføres 1, 2 og 5 år etter diagnose ved regionsykehusene i hht nasjonalt tverrfaglig oppfølgingprogram for barn med tumor cerebri, og det gjøres en vurdering av behov for utredning også tidligere i forløpet. Pedagogisk oppfølging ved behov er vesentlig.
- Habilitering og andre former for oppfølging individualiseres.
Høygradige gliomer og Diffuse ponsgliomer
Høygradige gliomer (HGG) er de vanligste CNS-svulster hos voksne, men betydelig sjeldnere hos barn. De aller fleste er astrocytomer, og etter oppdatert WHO- klassifiseringen av CNS svulster fra 2016 skilles det mellom anaplastisk astrocytoma IDH wild type eller IDH mutert (AA) (WHO grad 3), glioblastoma (GB) IDH wild type eller IDH mutert (WHO grad 4) og diffuse midtlinje gliomer, H3K27M-mutert (WHO grad 4) (Gupta & Dwivedi, 2017). Høygradige astrocytomer hos barn har en annen mutasjonsspektrum sammenlignet med voksne svulster. For eksempel viser anaplastiske astrocytomer hos barn veldig sjelden IDH mutasjon som er mer typisk hos voksne. Diffuse ponsgliomer (DIPG- Diffuse Intrinsic Pontine Glioma) er en undergruppe av diffuse midtlinje gliomer og omtales separat da de har egen behandling på grunn av spesifikk biologi og klinisk forløp med svært dårlig prognose.
Diffuse/Fibrillære astrocytomer (WHO grad 2) er lavgradige gliomer, men det kan være en vanskelig avgrensning mot grad 3, og svulstene kan utvikles i malign retning med tiden.
Vi finner HGG i hele CNS, fra 35–50 % supratentorelt, 40–50 % oppstår i hjernestammen inkludert ponsområdet som diffuse ponsgliomer(DIPG), i midtlinjestrukturer inkludert thalamus (13 %), i lillehjernen (5 %) og i spinalkanalen (3 %) (Gianno F, 2018).
Epidemiologi
HGG har en insidens på ca. 0,85 pr 100 000 barn og ungdom fra 0–19 år pr år og utgjør ca. 8–12 % av alle CNS-svulster hos barn (Braunstein, Raleigh, Bindra, Mueller, & Haas-Kogan, 2017).
Strålebehandling mot CNS fører på sikt til en økt risiko for utvikling av HGG.
Patologi
Både AA og GB vokser diffust infiltrativt og tumor kan være vanskelig å avgrense på MR bilder. Begge svulstene har høy mitotisk aktivitet, høy cellularitet, kjerneatypi og cellulær pleomorfisme. Hos GB finner vi i tillegg varierende områder med nekrose, småblødninger, fast og bløtt vev, i tillegg til mikrovaskulær proliferasjon.
Skillet mellom grad 3 (AA) og grad 2 (DA/FA) kan være vanskelig, spesielt i små biopsier, og patologens diagnostisering vil influere betydelig på resultatene i studier for hhv HGG og LGG etter hvordan grensetilfellene blir vurdert.
Molekylærbiologi
De siste årene har det vært en voldsom økning i kunnskapen om molekylærbiologiske forhold ved hjernesvulster og for HGG er det funnet stor heterogenitet. Kartlegging av arvematerialet ved HGG har bl.a. påvist mutasjoner som affiserer histonfunksjonen i svulstcellene (K27M og G34R/V mutasjon i H3.3 og H3.1), og som er et kjennetegn ved HGG hos barn og unge voksne. Videre er det funnet molekylære subgrupper (for.eks. H3.3 K27M, H3.1 K27M, H3.3 G34R/V) som kan si noe om svulstens plassering i CNS, typisk alder når svulsten opptrer og om prognose.
I kortikale svulster finnes ofte mutasjon eller overekspresjon av TP53, i tillegg til metylert MGMT og 5–10 % av svulstene i dette område utrykker BRAF V600E mutasjon.
Det finnes også andre genetiske forandringer som epidermal vekstfaktor-reseptor (EGFR) og platederivert vekstfaktor-reseptor A (PDGFRA) som kan være mutert og/eller ha amplifikasjon/overekspresjon i ulike sub-grupper av HGG (Sturm, Pfister, & Jones, 2017).
Diagnose og utredning
Som ved de fleste andre hjernesvulster avhenger symptomene ikke av histologi, men mest av tumors lokalisasjon som forårsaker lokal påvirkning eller økt intrakranielt trykk. Ofte har symptomene vart i kort tid pga. svulstenes aggressive vekst. Hodepine, kvalme/oppkast, epilepsi og hemiplegi er vanlige symptomer. Andre fokalt fremkalte symptomer forekommer. Rask forverring skjer hvis pasienten ikke blir behandlet.
Ved MR undersøkelse av HGG er tumor uklart begrenset med irregulære marginer. Som oftest ses inhomogen kontrastoppladning. GB viser oftest sentral nekrose omgitt av en hyperintens sone og ødem rundt tumor er vanlig.
Tumor er som oftest fokal, men kan gi spinal spredning, slik at hele CNS skal visualiseres.
Histopatologisk diagnose er helt nødvendig, og materiale fås ved reseksjon eller biopsi.
Behandling
Kirurgi er en svært viktig del av behandlingen, og målet er å fjerne hele svulsten radikalt hvis mulig, eventuelt så mye som mulig uten for stor skade. Da svulsten har et infiltrativt vekstmønster, vil det alltid være gjenværende tumorceller rundt reseksjonen.
Fokal strålebehandling gis til alle barn over 3 år. HGG har ikke mikrospredning til CNS slik at stråling av hele CNS er ikke nødvendig. Senskadene blir dermed mindre.
Kjemoterapi
Allerede i 1976 ble det i USA startet behandling med kjemoterapi bestående av prednison, lomustin og vincristin (pCV), med signifikant bedring av resultatene i forhold til stråling alene. Det samme ble vist i andre studier hvor kjemoterapi i tillegg til strålebehandling gav økt overlevelse i forhold til bare stråling (Sposto et al., 1989). Senere har det i flere studier vært ny histologisk gjennomgan av tumorvev som har vist at mange lavgradige gliomer har vært inkludert i studiene, og at kjemoterapi i tillegg til kirugi og stråling ikke gav bedre overlevelse ved HGG (Fouladi et al., 2003).
Senere utprøving av nye regimer har ikke klart å bedre resultatene for HGG. Temozolomid sammen med og etter strålebehandling har hatt en viss effekt hos voksne, men det er ikke funnet samme effekt hos barn(K. J. Cohen et al., 2011).
Det prøves ut en rekke nye stoffer og prinsipper i behandlingen av HGG hos barn. Lovende resultater har blitt oppnådd med vaksinasjon med dendritiske celler. Ut fra økende molekylær forståelse av HGG hos barn prøves det ut stoffer som påvirker forskjellige molekylære signalveier, i form av monoklonale antistoffer, småmolekylinhibitorer, strålesensitizere, genterapi og immunterapi. Disse kan påvirke celleproliferasjon, angiogenese, invasivitet og celleoverlevelse (Hargrave, 2009). Rebestråling av supratentoriale HGG har vist forlenget levetid sammenliknet med barn som ikke fikk rebestråling (Tsang, Oliveira, et al., 2019).
For øyeblikket er det ingen åpne studieprotokoller for barn med HGG. Internasjonalt er det ikke konsensus om beste behandling. De fleste barn i Norge får kombinasjonsbehandlingen med stråling og Temozolomid eller stråling og to ulike kjemoterapi, Temozolomid og Lomustine. Den siste kombinasjonen har vist seg å være mer effektiv enn Temozolomid alene, spesielt hvis tumorvevet utrykkeraktiv (umetylert) MGMT enzym promotor som medfører begrenset effekt av Temodal (Jakacki et al., 2016).
HGG hos barn < 3 år er veldig sjeldent og de har en bedre prognose sammenliknet med eldre barn, grunnet ulik biologi i svulstvevet (Gielen et al., 2015). Disse barna har nytte av kjemoterapi og anbefales behandling etter «SIOP Infant HGG protocol».
Prognose
Det har vært lite fremgang i resultatene for HGG. 5 års overlevelse ligger mellom 15 og 25 %. Histologi og omfanget av tumorreseksjon er viktige prognostiske faktorer, i tillegg til proliferasjonsindex. AA har bedre prognose enn GB og total tumorreseksjon og lavere proliferasjons index gir bedrer prognosen. I omtrent 40 % av HGG påvises mutasjon i tumor supressor genet, TP53, noe som er forbundet med dårlig prognose. Påvisning av mutasjoner i H3F3A, genet som koder for histonfunksjonen i svulstcellene, gir opphav til ulike molekylærbiologiske subgrupper som kan si noe om svulstens plassering i CNS, typisk alder når svulsten opptrer og om prognose. Svulster med H3.3 K27M mutasjon og H3.1K27M mutasjon finnes ofte i hjernestammen og har svært dårligprognose, mens H3.3 G34R/V mutasjon er cortikal svulster med bedre prognose (Braunstein et al., 2017).
BRAF V600E mutasjon finnes i 5–10 % ab HGG og er forbundet med bedre prognose. For disse pasientene finnes det «target therapy» i form av BRAF inhibitor som så langt har vist lovende resultater (Robinson, Orr, & Gajjar, 2014).
Diffuse ponsgliomer
Dette er gliomer som infiltrerer diffust i den sentrale hjernestamme og utgjør en egen klinisk tilstand, uavhengig av tumors histopatologi, som i de aller fleste tilfeller er astrocytom WHO grad 2, 3 eller 4. På engelsk kalles svulsten «Diffuse Intrinsic Pontine Glioma» (DIPG). Tumor sitter nesten alltid sentralt i pons, og diagnosen stilles på bakgrunn av klassiske MR-funn og spesifikke kliniske symptomer. Prognosen er særs dårlig, bare spredte langtids overlevere er beskrevet. Det er gjort mange behandlingsforsøk med forskjellige tilnærminger uten at noen har hatt tilfredsstillende effekt. Strålebehandling har en viss livsforlengende effekt.
Det er imidlertid viktig å være klar over at det også finnes mer fokale (ikke- diffuse) svulster i hjernestammen med et helt annet forløp. Disse er som oftest lavgradige gliomer med forløp og behandling som LGG andre steder. Prognosen ved LGG i hjernestammen varierer, men kan være ganske god med høy langtidsoverlevelse. (Bemerk at fibrillært astrocytom (WHO grad 2) som manifesterer seg som DIPG ikke regnes som LGG.)
Epidemiologi
Svulster i hjernestammen utgjør 10–15 % av alle pediatriske CNS svulster hos barn og DIPG utgjør 80 % av disse. DIPG forekommer i alle aldersgrupper, men er hyppigst å alderen 5–10 år (Vanan & Eisenstat, 2015).
Patologi
Tidligere var det ikke vanlig å biopsere DIPG, både på grunn av svulstens beliggenhet i et sårbart område og begrenset terapeutiske muligheter. Muligheten for å finne molekylære endringer i svulstvevet og påfølgende målrettet terapi, har ført til en økt biopsering av DIPG, noe som har vist seg å være relativt trygt. Den pågående BIOMEDE studien for DIPG hos barn og ungdommer, krever biopsi for inklusjon i studien.
Nesten alle biopserte svulster viser astrocytom WHO grad 2–4 og fordelingen mellom disse varierer i publiserte materialer, men forløpet varierer ikke mellom de forskjellige histopatologiske typene. Det er beskrevet enkelte DIPG med PNET histopatologi.
Molekylærbiologi
Den økende biopsering ved DIPG har vist at over 80 % –90 % av DIPG innehar H3K27M mutasjon som affiserer histonfunksjon i svulstcellene (Sturm et al., 2017). Dette er bakgrunnen for den nye histologiske diagnosen i siste WHO-utgave (Gupta & Dwivedi, 2017) . Denne mutasjonen finnes også i andre høygradige midtlinjegliomer.
Diagnose og utredning
På grunn av sykdommens aggressive biologi, inntrer symptomene akutt og progredierer raskt. Oftest har symptomene vart mindre enn 2–3 måneder ved diagnose. Pasientene har klassiske hjernestammesymptomer som abducensparese, facialisparese, dysfagi, hemiplegi og ataksi.
MR cerebrum gir diagnosen. Svulsten utvider pons og infiltrerer diffust omgivelsene langs fibertraktene. Den gir betydelig masseeffekt med sammenklemming av fjerde ventrikkel, men mindre enn 10 % har hydrocefalus ved diagnose. Tumor er hypointens på T1, hyperintens på T2 og FLAIR. Den tar sjelden kontrast. Det siste er viktig differensialdiagnostisk i forhold til pilocytisk astrocytom.
DIPG diagnostiseres radiologisk og er alltid inoperabel. Biopsi av DIPG er blitt mye vanligere, grunnet muligheten for å finne molekylære endringer i svulstvevet og påfølgende målrettet terapi,
Ved ikke-diffuse hjernestammesvulster og når det er tvil om diagnosen DPG, skal det gjøres biopsi.
Behandling
Kirurgi. DIPG er alltid inoperabel. Vedrørende biopsi, se ovenfor.
Strålebehandling er standardbehandling og den eneste behandling med dokumentert effekt ved DIPG. Effekten er forbigående klinisk bedring og opptil 3 mnd. forlengelse av levetiden. Vanlig dose er 54 Gy til tumorområdet. Det gis ikke stråling til hjernen for øvrig. Behandlingen er ikke kurativ. Disse pasientene er i øyeblikket ikke kandidater for protonstrålebehandling. Re-bestråling har vist økt overlevelse, sammenliknet med standardbehandling (Lu, Welby, Mahajan, Laack, & Daniels, 2019).
Kjemoterapi og annen behandling. De fleste cytostatika i varierende kombinasjoner og doser har blitt prøvd kombinert med strålebehandling, uten bedre effekt enn strålebehandling alene. Høydosebehandling har heller ikke bedret resultatene, heller ikke Angiocomb, en studie som kombinerte topotecan som radiosensitizer gitt under strålebehandlingen, etterfulgt av kurer med de anti-angiogenetiske medikamentene thalidomid og celecoxib, samt etoposid. Utprøving med stoffer som gemcitabine (nukleosid analog), gefitinib (EGFR inhibitor), tipifarnib (farnesyltransferase inhibitor) og flere tyrosin kinase hemmere, som påvirker molekylære prosesser i svulsten, har heller ikke vist bedre resultater. I Norge (2019) inkluderer vi pasienter i BIOMEDE (Biological Medicine for Diffuse Intrinsic Pontine Glioma (DIPG) Eradication (BIOMEDE), 2014-), som er en europeisk behandlings-protokoll for DIPG. Etter biopsi fra tumor, blir alle bestrålt, i tillegg til biologisk medisin som styres etter svulstens biologiske karakter (Everlimus (mTOR inhibitor), Erlotinib eller dasatinib (begge tyrosin kinase hemmer). Resultatene fra BIOMEDE forventes i løpet av 2020.
Prognose
Prognosen er svært dårlig med median overlevelse på 9–13 måneder. Dette har ikke endret seg de senere år. Overlevelsestall varierer i litteraturen, ofte kan det være uklart om angitte overlevere egentlig har en annen type hjernestammesvulst. Det er imidlertid påvist enkeltpasienter med godt dokumentert DIPG som overlever lenge.
Det er ikke beskrevet prognostiske faktorer som har behandlingsmessige konsekvenser, men kort sykdomsvarighet og betydelige nevrologi ske symptomer har en viss negativ prognostisk betydning. Lavere malignitetsgrad (WHO grad 2) har ikke innflytelse på forløpet.
Ependymom
Ependymom er den tredje vanligste hjernesvulsten hos barn og utgjør 8–12 % av alle hjernesvulstene hos barn og ungdom. Mer enn halvparten av tilfellene opptrer hos barn under 5 år og vi antar at svulsten utgår fra ependymcellene som dekker hjernens ventrikler og sentralkanal i spinalkanalen (Khatua, Ramaswamy, & Bouffet, 2017).
Lokalisering
Ependymomene er stort sett lokalisert i tilknytning til ventriklene, men kan forekomme i hele sentralnervesystemet (CNS), uavhengig av og langt fra områder med ependymceller. Det forklares ved transformasjon av embryologiske rester av ependymceller inne i hjerneparenchymet eller en transformasjon av nevrale stamceller (Reni, Gatta, Mazza, & Vecht, 2007; Zacharoulis & Moreno, 2009). Hos barn er 90 % av ependymomene intrakranielle og av disse er 2/3 lokalisert i bakre skallegrop i tilknytning til 4. ventrikkel (Khatua et al., 2017).
Histopatologi og molekylærbiologi
Etter oppdatert WHO- klassifiseringen av CNS svulster fra 2016 skilles det mellom WHO grad I subependymom og myksopapillær ependymom, som hovedsakelig opptrer hos voksen, WHO grad II ependymom og WHO grad III anaplastisk ependymom (Gupta & Dwivedi, 2017). For WHO grad II ependymomer finnes tre histologiske varianter: pappillært, klarcellet og tancytisk ependymom. RELA fusjon positiv ependymom er en molekylær variant av supratentorielle ependymomer og kan enten være WHO grad II eller WHO grad III. De vanligste histopatologiske subtypene hos barn er klassisk og anaplastisk ependymom. Den klassiske typen viser perivaskulære pseudorosetter av gliale tumorceller uten tydelig tegn til anaplasi, mens den anaplastiske typen viser forhøyet mitotisk aktivitet, mikrovaskulær proliferasjon og utbredte nekroser. Det er glidende overganger mellom de to typene og ofte vanskelig å avgjøre hvilken gruppe de tilhører (Ellison et al., 2011). Det er foreløpig ingen pålitelige biologiske prognostiske markører for ependymom, og sammenhengen mellom det histopatologiske bildet og prognose har ikke vist seg å gjenspeile seg i kliniske studier (Ellison et al., 2011; Lundar, Due-Tonnessen, Scheie, & Brandal, 2014).
De siste årenes økning i kunnskapen om molekylærbiologiske forhold ved hjernesvulster har vist en stor heterogenitet når det gjelder ependymom og det er funnet 9 molekylære subgrupper av ependymale svulster, 3 for hvert hovedområde i hjernen: supratentorielt, bakre skallegrop og spinakanal. For supratentoriale ependymom (ST-EPN) hos barn er anaplastisk EPN med YAP1-fusjon og anaplastisk EPN med RELA-fusjon de vanlige subgruppene, hvor YAP1-fusjon er forbundet med svært god prognose og RELA-fusjon langt dårligere prognose. For ependymom i bakre skallegrop (PF-EPN) er det også hovedsakelig 2 subgrupper hos barn, anaplastisk gruppe A med dårlig prognose og anaplastisk gruppe B med god prognose. I spinalkanalen er det funnet 3 subgrupper, men alle er forholdsvis sjeldne hos barn (Pajtler et al., 2015). Forekomsten av tillegg av kromosom 1q indikerer dårlig prognose og stor sannsynlighet for tilbakefall.
Diagnose og utredning
De vanligste symptomene ved ependymom er spesifikke og relatert til økt intrakranielt trykk fra obstruktiv hydrocephalus som morgenhodepine, brekninger om morgenen og evn. personlighetsforandringer, men nevrologiske utfall og cerebellare symptomer som ataksi kan forekomme. Hos små barn kan apati og irritabilitet være de eneste symptomene. Diagnosen stilles ved MR-undersøkelse av hele CNS-aksen, undersøkelse av spinalvæsken og histologisk undersøkelse av svulstvevet.
Behandling
Behandling av ependymom er som oftest multimodal og krever høyspesialiserte, bredt sammensatte nevro-onkologiske team. Den gjennomføres i henhold til internasjonale studieprotokoller/retningslinjer.
Kirurgi
Standard behandling av ependymom er først og fremst kirurgi, etterfulgt av fokal strålebehandling (Merchant et al., 2009). Kjemoterapi har en mer usikker effekt på svulstvevet (Bouffet, Tabori, Huang, & Bartels, 2009). Den optimale behandlingen vil være total tumorfjerning, men ofte er det vanskelig grunnet svulstens evne til å infiltrere vitale strukturer, spesielt i bakre skallegrop. Målet for kirurgisk behandling vil være maksimal tumorfjerning, samtidig med et optimalt nevrologisk resultat. Studier har vist at det som betyr mest for overlevelsen, er grad av resttumor. Det har den senere tid ført til en mer aggressiv kirurgisk tilnærming med «second look» kirurgi for å oppnå totalreseksjon av tumor, og dermed bedre overlevelsen (Merchant et al., 2009). Studier har vist at det er gjennomførbart uten alvorlig morbiditet (Massimino et al., 2004).
Strålebehandling
Standard behandling av ependymom er kirurgi, etterfulgt av strålebehandling hos større barn og ungdom og kjemoterapi hos de minste barna som ikke kan få strålebehandling. Studier har vist at behandling med både kirurgi og strålebehandling har bedre overlevelse (Schroeder et al., 2008; Swanson et al., 2011) Protonstråling bør være standard behandling.
CNS-metastaser forekommer, men i langt mindre frekvens enn man tidligere trodde og derfor er det kun tumorområdet med marginer som blir bestrålt. Dosen må over 54 Gy og helst så høyt som 59.4. I den nyeste behandlingsprotokollen SIOP Ependymoma II er stråledosen 59,4 Gy for barn > 18 mnd., med mindre stråledoser (54 Gy) til barn mellom 12–18 mnd. og til barn som har gjennomgått flere kirurgiske inngrep (An International Clinical Program for the Diagnosis and Treatment of Children With Ependymoma (SIOP-EP-II), 2015-2026).
For ependymom i hjernestammen er det flere land, inkludert Norge, som har vært skeptisk til en så høy stråledose (59,4 Gy) mot et sårbart område og påfølgende fare for strålenekrose.
Cytostatika
Cytostatikabehandling ved ependymom har usikker effekt og beste standard behandling er ikke etablert (Bouffet et al., 2009). Cytostatika brukes på de minste barna i påvente av strålebehandling og cytostatika har trolig en viss effekt (Grundy et al., 2007). Andre studier viser ikke den samme effekten (Grill et al., 2001).
I «SIOP Ependymoma II» får alle spedbarn < 12 måneder og barn med resttumor, cytostatika. I protokollen inngår cyclofosfamid, vincristin, carboplatin, etoposid, cicplatin og metotrexate i forskjellige kombinasjoner. I tillegg blir spedbarn < 1 år eller barn som ikke har fått strålebehandling, randomisert til Valporat eller ikke i vedlikeholdsbehandlingen.
Barn med ependymom i Norge behandles i utgangspunktet etter «SIOP Ependymoma II», men med visse modifikasjoner. Stort sett vil en i Norge gå for mindre stråling enn anbefalt i protokollen når tumor ligger i hjernestammen. Faggruppen for CNS svulster hos barn har bestemt at Norge skal delta i «SIOP Ependymoma II», og arbeidet med å åpne studien i Norge er allerede i gang.
Prognose
Prognosen for ependymom er relativt dårlig med 5 års PFS mellom 23–74 % og OS mellom 60 %–80 % (Cage et al., 2013). Den varierer med alder, WHO-klassifikasjon, lokalisasjon og resttumor etter kirurgi. Det som har betydd mest prognostisk er grad av resttumor etter kirurgi og ved komplett reseksjon av WHO grad 1–3 er det > 60 % 5-års overlevelse (Pajtler et al., 2017; Pejavar et al., 2012). Overlevelse basert på de nye molekylære subgruppene av ependymale svulster, viser at ST anaplastisk EPN med YAP1-fusjon og PF anaplastisk EPN gruppe B har svært god prognose (10 års OS 88–100 %), mens ST anaplastisk RELA-fusjon og PF anaplastisk EPN gruppe A har langt dårligere prognose (10 års OS 50 %) (Pajtler et al., 2015).
Oppfølging
Alle barn kontrolleres med klinisk undersøkelse og MR av hele CNS hver 3. mnd. første året etter avsluttet behandling, deretter hver 4. mnd. andre året og videre halvårige kontroller frem til 5 år etter avsluttet behandling. I tillegg følges alle barn minst 1 gang årlig med tanke på sekveler av behandlingen. Mange barn med ependymom vil i varierende grad få senfølger av behandlingen. Dette må følges nøye. Nevropsykologisk kartlegging gjennomføres på alle 1, 2 og 5 år etter diagnose i hht nasjonalt tverrfaglig oppfølgingsprogram, og det gjøres en vurdering av behov for dette også tidligere i forløpet. Pedagogisk oppfølging ved behov er vesentlig. Videre oppfølging av barnenevrolog, fysioterapeut, endokrinolog, nyrefunksjon, hørsel, ØNH-status og øyestatus gjøres i varierende grad. Der bivirkninger av behandlingen påvises, etableres det et samarbeid med lokale aktører for å legge til rette for best mulig habilitering
Residivbehandling
Residiv ved ependymom forekommer hos 30–50 % av pasientene og er forbundet med veldig dårlig prognose (Zacharoulis et al., 2010). Det finnes ingen standard behandling. Nytt operativt inngrep må alltid vurderes og der hvor strålebehandling ikke er gjennomført, må det vurderes. Kraniospinal re-bestråling har vist økt overlevelse sammenliknet med fokal re-strålebehandling, mens cytostatika har vist dårlig effekt (Tsang, Murray, et al., 2019). Utprøvende behandling med biologisk medisin vil mulig gi større håp for denne pasientgruppen i fremtiden.
Medulloblastom
Medulloblastom er en malign svulst av embryonal opprinnelse lokalisert i lillehjernen, som opptrer særlig hos barn. Det er en biologisk heterogen svulstgruppe, og det er nå bred enighet om at det eksisterer 4 molekylærgenetiske subgrupper med betydning for prognose. I WHOs klassifikasjon av svulster i sentralnervesystemet fra 2016 er disse nå tatt med, i tillegg til de histologiske undergruppene av medulloblastom (Louis et al., 2016).
Symptomene er enten sekundært til svulstens tendens til å gi vannhode/ hydrocephalus eller direkte påvirkning av (lille)hjerne eller ryggmarg. Kvalme, brekninger, trunkal ataxi og koordinasjonsproblemer er de vanligste symptomene.
Diagnose
MR-undersøkelse som fremstiller hjerne og ryggmarg er nødvendig og gir mistanke om diagnosen, men endelig diagnose og klassifisering kan ikke bestemmes før etter histologisk og molekylærbiologisk undersøkelse av svulstmaterialet og spinalvæske.
WHOs klassifikasjon av medulloblastomer (2016)
Medulloblastomer klassifiseres under embryonale tumores og er høygradig maligne (WHO grad IV). Følgende undergrupper er definert:
Både molekylærbiologisk og histologisk definert medulloblastom brukes når diagnosen stilles, og de ulike kombinasjonene av genetiske og histologiske undergrupper har ulik prognose og forekommer i ulike aldersgrupper. Se tabell under:
Table 5: Summary of the most common integrated medulloblastoma diagnoses, with clinical correlates
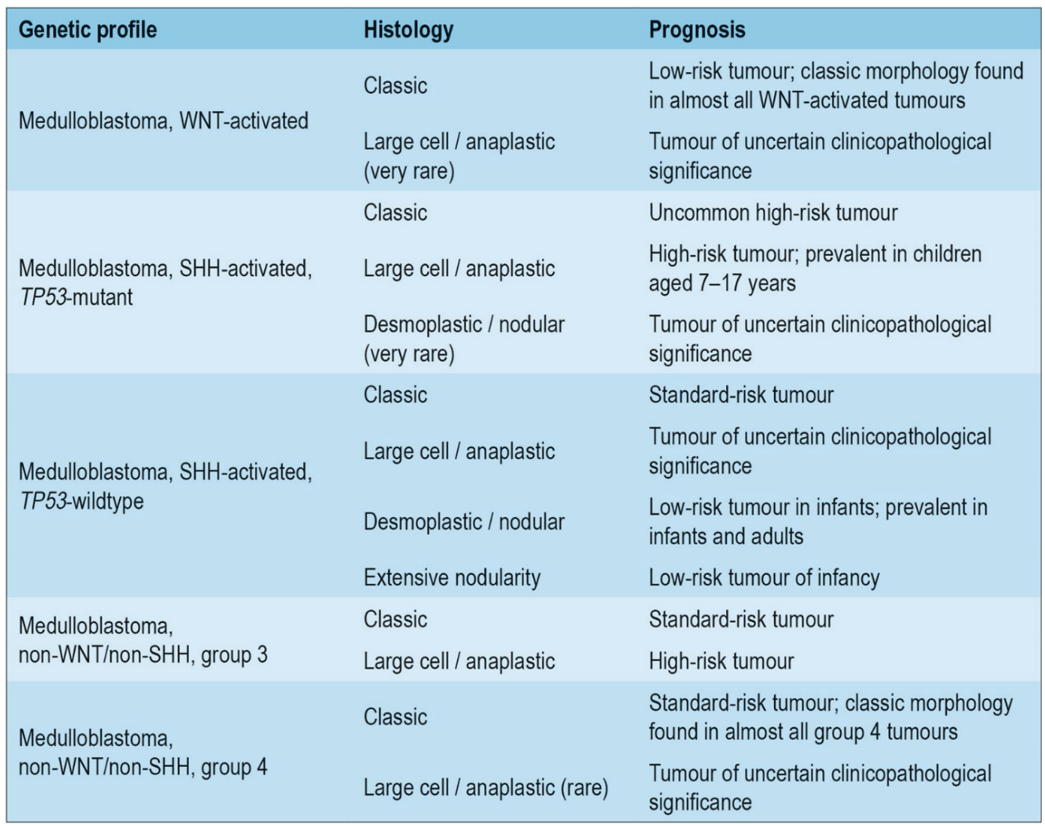
Molekylærbiologiske markører
Tumormateriale vil bli undersøkt på følgende markører for å evaluere prognostisk betydning: CTNNB1-mutasjoner (WNT-aktiverte medulloblastomer), APC-mutasjoner (WNT-aktiverte CTNNB1-villtype medulloblastomer), monosomi 6 (WNT-aktiverte medulloblastomer), gain/amplifikasjon av C-MYC og N-MYC, TP53-mutasjoner, PTCH1-mutasjoner, og SUFU-mutasjoner (SHH-aktiverte medulloblastomer).
Flere av disse markørene er implementert som prognostiske faktorer i behandlingsprotokoller.
Stadieinndeling (Chang)
M0: | Lokalisert til lillehjernen uten tegn til lokal spredning eller spredning til hjernehinnevæsken |
M1: | Spredning til hjernehinnevæsken |
M2: | Spredning lokalt i lillehjernen |
M3: | Spredning til storehjerne/ryggmargen |
M4: | Spredning utenfor sentralnervesystemet |
Standard risk: | M0 og postoperativ resttumor under 1.5 cm2 |
Høyrisk: | M1–4, storcellet eller anaplastisk histologi, og/eller MYC-amplifisert |
Behandling
Kirurgi alene er ikke kurativt ved medulloblastom. Adjuvant behandling er helt nødvendig for overlevelse. Barn over 4 år vil rutinemessig bli behandlet med strålebehandling, helst protonbestråling, mot hjerne og ryggmarg. Barn under 4 år vil bli behandlet med kjemoterapi som eneste adjuvante behandling. Opptil 1/3 av pasientene har spredning til hjernehinnene ved diagnose. Valg av behandlingsprotokoll og -stratifisering vurderes og bestemmes i MDT-møte.
Kirurgi er meget viktig for prognosen da minimal restsykdom etter kirurgi gir best prognose. Ved restsvulst på mindre enn 1,5 cm2 på postoperative MR-bilder ser man en bedre prognose/overlevelse enn ved større resttumor. Som for de fleste andre svulsttyper vil derfor målsetningen med kirurgi ved medulloblastom være maksimal svulstreseksjon uten å løpe for stor risiko for å påføre pasienten nevrologiske sekveler.
Stråleterapi er en viktig del av behandlingen og hele nevralaksen skal bestråles med boost mot primærtumorområdet og eventuelle metastaser. Dose til nevralaksen varierer fra 18 til ca 36 Gy, mens man til primærtumorområdet og eventuelle metastaser totalt gir 54 Gy (ca 50 Gy ved bestråling spinalt). Grunnet risiko for alvorlige senskader anbefales ikke bestråling til de minste barna. Stråling med protoner er standardbehandling i dag. Mer spesifikt gir man for norske barn under 4 år ikke strålebehandling til nevralaksen, mens man vurderer om det er aktuelt med lokal bestråling mot tumortomt og eventuell resttumor for de som er fra 2 år og oppover. Ett av ankepunktene mot sistnevnte strategi er at det ved recidiv blir vanskeligere å gi ny strålebehandling.
Cytostatikabehandling er under kontinuerlig evaluering. Medulloblastomer er vanligvis kjemoterapisensitive, men beste standardbehandling er ikke endelig fastslått. Hos barn < 4 år startes det med postoperativ cytostatikabehandling i håp om å utsette stråleterapi lengst mulig (Rutkowski, 2006; Rutkowski et al., 2005). Hos noen spesifikke undergrupper av medulloblastom hos de aller yngste barna, er det også god prognose ved kun adjuvant behandling med cytostatika, inkludert methotrexate intratekalt.
Standard risk < 4 år: | HIT MED Guidance |
Standard risk > 4 år: | SIOP PNET 5 |
Høyrisk alle aldre: | HIT MED Guidance |
Cytostatika som inngår er cyclofosfamid, vinkristin, metotrexat, karboplatin, etoposid, cisplatin og CCNU i varierende kombinasjoner og intensitet alt etter alder og sykdomsutbredelse. Mange av høyrisk-pasientene og de under 4 år får også intratekal metotrexat. Alle barn over 4 år får bestråling mot CNS-akse. Barn under 4 år behandles rutinemessig med kun cytostatika.
Prognose
Best prognose har standard risk pasienter med ca 70 % langtidsoverlevelse, mens prognosen for de yngste barna (< 4 år) er dårligere. Høyriskpasientene > 4 år har ca 40–50 % langtidsoverlevelse, og også blant disse er det dårligst prognose for de minste barna (< 4 år).
Oppfølging
Alle barn kontrolleres og diskuteres i MDT-møter. Rutinemessig billedoppdatering og tumorkontroll etter 3, 6, 12 18 og 24 mnd med MR, siden MR hvert år til 5 år etter avsluttet behandling. Etter 5 år er det individuell oppfølging, men pga at de fleste har fått strålebehandling mot total CNS-akse, vil vi anbefale MR-kontroller livslangt.
I tillegg bør alle barn følges minst 1 gang årlig med tanke på sekveler av behandlingen. Alle barn med medulloblastom vil i varierende grad få senfølger av behandlingen. Nevropsykologisk kartlegging gjennomføres for alle 1, 2 og 5 år etter diagnosetidspunkt i hht nasjonalt tverrfaglig oppfølgingprogram for barn med tumor cerebri og det gjøres en vurdering av behov for dette også tidligere i forløpet. Pedagogisk oppfølging ved behov er vesentlig. Videre oppfølging av barnenevrolog, fysioterapeut, endokrinolog, nyrefunksjon, hørsel, ØNH-status og øyestatus gjøres i varierende grad. Der bivirkninger av behandlingen påvises, etableres det et samarbeid med lokale aktører for å legge til rette best mulig habilitering.
Residivbehandling
Ved residiv er behandlingsresultatene nedslående. De terapeutiske mulighetene er i de fleste tilfellene brukt i første omgang. Der hvor strålebehandling ikke er gjennomført, må det nå vurderes. Reoperasjon kan være aktuelt. Førstevalget i Norge for barn med residiv av medulloblastom, er Memmat-protokollen. Denne protokollen er nå åpen for inklusjon av norske pasienter. Dette er en metronomisk og målrettet anti-angiogenetisk behandlingsprotokoll, som også inkluderer intratekal cellegift.
Supratentoriell PNET
Andre embryonale hjernesvulster
Disse svulstene gikk tidligere under fellesbetegnelsen supratentoriell PNET (primitiv nevroektodermal tumor). Medulloblastom er en primitiv nevroektodermal tumor lokalisert i lillehjernen, mens disse andre ble kalt supratentoriell PNET da de var lokalisert supratentorielt. PNET er imidlertid ikke lenger en egen gruppe svulster i WHO-klassifiseringen fra 2016, men er nå delt opp i flere ulike entiteter i gruppen embyonale svulster (Louis et al., 2016).
Uspesifikke symptomer som tegn på økt intrakranielt trykk, kramper og nevrologiske utfall er det som oftest bringer pasienten til kontakt med helsevesenet.
Diagnosen stilles ved MR-undersøkelse. Hele CNS-aksen må visualiseres og vevsprøve fra svulsten samt cytologi fra spinalvæsken må undersøkes for endelig diagnose og klassifisering.
WHOs klassifikasjon av embryonale svulster som ikke er medulloblastomee (2016) inneholder følgende undergrupper:
- Embryonal tumor med rosetter, C19MC-endret (ETMR)
- Embryonal tumor med rosetter, NOS (ETMR)
- Medulloepiteliom
- CNS nevroblastom
- CNS ganglionevroblastom
- CNS embryonal svulst, NOS
- Atypisk teratoid/rhabdoid tumor (se eget kapittel)
- CNS embryonal tumor med rhabdoide trekk
Molekylærbiologiske markører
Tumormateriale vil bli undersøkt med tanke på mange molekykærbiologiske markører, noe avhengig av hvilken type embryonal svulst man mistenker. For eksempel vil CNS nevroblastom oftest ha gevinst av kromosomarm 1p, tap av kromosomarm 16q og FOXR2-forandringer,
Behandling
Behandling av disse embryonale svulstene krever høyspesialiserte og bredt sammensatte nevroonkologiske team (MDT). Behandlingen gjennomføres i henhold til internasjonale studieprotokoller/retningslinjer (B. H. Cohen et al., 1995; Massimino et al., 2006; Timmermann et al., 2006).
Kirurgi er meget viktig for prognosen da minimal restsykdom etter kirurgi gir best prognose. Operasjoner bør derfor gjøres der det finnes erfarne nevrokirurger med omfattende erfaring med barn og god barneanestesi-/intensivekspertise.
Stråleterapi er en viktig del av behandlingen og for å ha et realistisk håp om kurasjon må hele nevralaksen bestråles med boost mot primærtumorområdet og eventuelle metastaser. Dose til nevralaksen vil for de fleste være ca 36 Gy, mens man til primærtumorområdet og eventuelle metastaser totalt gir 54 Gy (ca 50 Gy ved bestråling spinalt). Grunnet risiko for alvorlige senskader anbefales ikke bestråling til de minste barna (Timmermann et al., 2006). Mer spesifikt gir man for norske barn under 4 år ikke strålebehandling til nevralaksen, mens man vurderer om det er aktuelt med lokal bestråling mot tumortomt og eventuell resttumor for de som er fra 2 år og oppover. Ett av ankepunktene mot sistnevnte strategi er at det ved recidiv blir vanskeligere å gi ny strålebehandling. Stråling med protoner bør alltid vurderes.
Cytostatikabehandling er under kontinuerlig evaluering. Pasientene behandles oftest i henhold til HIT MED Guidance. Hos barn < 4 år startes det med postoperativ cytostatikabehandling i håp om å utsette stråleterapi lengst mulig.
Prognose
Prognosen varierer avhengig med type embryonal svulst og er vesentlig dårligere enn for medulloblastom. For hele gruppen (som tidligere ble benevnt PNET) ligger langtidsoverlevelsen ligger på ca 30–40 % (B. H. Cohen et al., 1995).
Oppfølging
Alle barn kontrolleres for tumorstatus etter 3, 6, 12 18 og 24 mnd med MR, siden MR hvert år til 5 år etter avsluttet behandling. Etter 5 år er det individuell oppfølging, men pga at de fleste har fått strålebehandling mot total CNS-akse, vil vi anbefale MR-kontroller livslangt.
I tillegg følges alle barn minst 1 gang årlig med tanke på sekveler av behandlingen. I varierende grad gjøres nevropsykologisk og spesialpedagogisk testing, undersøkelse av barnenevrolog, fysioterapeut, endokrinolog, nyrefunksjon, hørsel, ØNH-status og øyestatus. Der bivirkninger av behandlingen påvises, etableres det et samarbeid med lokale aktører for å legge til rette best mulig habilitering. Alle barn med PNET vil i varierende grad får senfølger av behandlingen som må følges nøye, og spesielt må endokrinologisk substitusjonsbehandling og pedagogisk tilrettelegging etableres.
Residivbehandling
Residivbehandling er nedslående. Der hvor strålebehandling tidligere ikke er gjennomført, må det nå vurderes. Reoperasjon kan være aktuelt. Utprøvende behandling med cytostatika kan startes, men resultatene så langt er ikke oppløftende.
Atypisk teratoid-rhabdoid tumor/ATRT
Atypisk teratoid / rhabdoid tumor beliggende i CNS (CNS AT/RT) er en svært ondartet svulst som hovedsakelig forekommer hos barn under tre år.
Diagnosen stilles ved histologisk undersøkelse.
Billedundersøkelse: Hele CNS-aksen må visualiseres og spinalvæsken undersøkes for klassifisering og utbredelse. Billedundersøkelse viser ofte cyster eller blødninger. På T1-vektet MR er svulstene hypointense, mens T2-vektede bilder viser iso- til hyperintense-lesjoner. Det er heterogen kontrastladning og ved leptomeningeal sykdom ser man ved billedundersøkelse en nodulær, klumpete kontrastoppladning langs meningene i spinalkanalen og inn i cauda equina. Billedundersøkelse alene kan ikke sikkert skille mellom AT/RT og suptratentorielle nevroblastomer (tidligere PNET) eller medulloblastom. Omtrent to tredjedeler av AT/RT-er forekommer i lillehjernen, vanligvis i den cerebellopontinvinkel, med infiltrasjon av omgivende strukturer. Omtrent en fjerdedel er supratentorial og 8 prosent er multifokale.
Patologi: Histologisk er AT/RT preget av rhabdoide celler og ligner på andre små runde blåcelletumorer. AT/RT kan imitere flere forskjellige entiteter; også medulloblastomer Nekrose og en høy grad av mitotisk aktivitet er vanlig. Germcellemarkører er negative.
Biologiske markører: Diagnostisering av AT/RT krever tap av enten INI1 kjernefarging, noe som indikerer biallel inaktivering av SMARCB1 på kromosom 22, eller tap av BRG1-farging, noe som indikerer SMARCA4 inaktivering. I tillegg til disse somatiske mutasjonene påvist i tumorvev, har omtrent en tredjedel av pasientene kimcelle mutasjoner i SMARCB1 eller, mindre vanlig, SMARCA4. Vanligvis er de minst delvis vimentin, CK, EMA og smooth muscle actin (SMA) positive. Ki67 er vanligvis relativt høy. Med hjelp av OTX2 fargning er det mulig å undersøke om det dreier om en AT/RT-Tyr. /flere av disse markørene vil kunne bli implementert som prognostiske faktorer i fremtidig protokoller (Japp, Klein-Hitpass, Denkhaus, & Pietsch, 2017; Johann et al., 2016).
Epidemiologi
AT/RT er den mest vanlige ondartede CNS tumores hos barn under 1 år, og AT/RT representerer omtrent 1–2 % hjernesvulster hos barn. I Norge har det vært 19 tilfeller av AT/RT i de 15 årene 2002–2017, det tilsier i snitt drøyt en pasient i året i hele Norge.
Symptomer: Symptomer hos spedbarn er ofte slapphet, oppkast og eller økende hodeomkrets; eldre barn kan ha symptomer grunnet affeksjon av hjernenervene IV, VI eller VII, for eksempel torticollis, dobbeltsyn eller facialisparese. Hemiplegi og/eller hodepine generelle trykksymptomer kan også være debutsymptomer.
Prognosen: I de fleste studier og rapporter er dårlig, med en median overlevelse på 12 til 14 måneder og fem års samlet overlevelse mindre enn 50 prosent. Pasienter med familiemutasjoner i SMARCB1genet har en tendens til å være yngre og ha dårligere prognose sammenlignet med de med ikke familiære, sporadiske svulster. Faktorer som er assosiert med bedre prognose inkluderer supratentoriell beliggenhet, ikke spredning av sykdommen ved diagnosetidspunktet, fullstendig reseksjon av tumor kirurgisk og uttrykk for ASCL1, en regulator for NOTCH-signalering.
Behandling: Kirurgi alene er ikke kurativt ved AT/RT, men en kombinasjon av kirurgi, omfattende cellegiftbehandling og strålebehandling er helt nødvendig for overlevelse (Fossey et al., 2017; Richardson, Ho, & Huang, 2018; Schrey et al., 2016).
Kirurgi er meget viktig for prognosen da minimal restsykdom etter kirurgi gir best prognose. Målsetningen med kirurgi ved AT/RT som ved andre ondartede CNS svulster er maksimal reseksjon.
Cytostatikabehandling er under kontinuerlig evaluering, og til nå har vi fulgt en anbefaling gitt i «A multinational registry for rhabdoid tumors of any anatomical site EUROPEAN RHABDOID REGISTRY, EU-RHA». AT/RT registeret er en anbefaling både når det gjelder utredning, behandling og oppfølgning av AT/RT. Her anbefales det å gi 9 kurer med kjemoterapi hver 2. uke, dels intravenøst og dels også intrathekalt i Ommaya reservoir. AT/RT svulstene er vanligvis initialt kjemosensitive. Alle barn får cytostatika under strålebehandling, men kurene modifiseres under strålebehandlingen på grunn av fare for økt toksisitet. I anbefalingene som er gitt i dette registret, har man også foreslått høydosebehandling med stamcellestøtte. Til nå har ikke høydosebehandling vist noen signifikant bedret overlevelse enn de 9 regulære kjemoterapikurer. Etter strålebehandling skal man ikke lenger gi intrathekal kjemoterapi.
Stråleterapi er en viktig del av behandlingen for å ha et realistisk håp om kurasjon, og man anbefaler å komme i gang med strålebehandlingen så tidlig som mulig i forløpet, i løpet av de første kurene. Ved ung alder og ikke dissiminert sykdom, anbefales lokal stråling mot tumorsengen, men ved eldre alder, resttumor og disseminert sykdom, anbefales å stråle hele nevralaksen med boost mot primærtumorområdet og eventuelle metastaser. Grunnet risiko for alvorlige senskader anbefales ikke CNS akse bestråling til de minste barna. Man anbefaler stråling med protoner.
Videre studier/planlagt behandling: Det er en planlagt protokoll med stratifiseringer og randomiseringer på trappene, men den kommende AT/RT protokoll er ikke offisiell ennå. Den vesentlige endringen er å gi 12 runder kjemoterpi i stedet for 9, : 3 dox, 4 VCA og 5 ICE.
Oppfølging: Alle barn kontrolleres med tanke på tumorkontroll etter 3, 6, 12 18 og 24 mnd med MR, deretter MR minimum to ganger årlig fra 2 til 5 år etter avsluttet behandling, og årlig til 10 år etter ferdig behandling. De barna som ikke har fått strålebehandling, anbefales å ta spinalpunksjon 2 ganger i året i 2 år etter ferdig behandling.
I tillegg følges alle barn minst 1 gang årlig med tanke på sekveler av behandlingen. Alle barn med AT/RT vil i varierende grad få senfølger av behandlingen som må følges nøye. Nevropsykologisk testing gjennomføres 1, 2 og 5 år etter diagnose i hht nasjonalt tverrfaglig oppfølgingprofgram for barn med tumor cerebri og det gjøres en vurdering av behov også tidligere i forløpet. Pedagogisk oppfølging ved behov er vesentlig. I varierende grad får barnet oppfølging av barnenevrolog, fysioterapeut, endokrinolog, nyrefunksjon, hørsel, ØNH-status og øyestatus. Det er en målsetting at der bivirkninger av behandlingen påvises, etableres samarbeid med lokale aktører for å legge til rette best mulig habilitering.
Residivbehandling: Ved residiv er behandlingsresultatene nedslående. De terapeutiske mulighetene er i de fleste tilfellene brukt i første omgang. Der hvor strålebehandling ikke er gjennomført, må det nå vurderes. Reoperasjon er sjelden aktuelt, da residivet ofte viser rask vekst, og tumor ikke lar seg fjerne. MEMMAT protokoll med metronomisk behandling med blant annet Avastin, peroral kjemoterapi, intrathekal behandling og diverse perorale medisiner (Celecoxib, Fenofibrat, Taidomid) kan vurderes. Målrettede terapier er også under utredning i en rekke fase-1 studier, og et stort antall medikamenter har potensiell terapeutisk aktivitet. Spesielt Aurora kinase A-hemmeren Alisertib, (MLN8237) har vist antitumoraktivitet hos barn med tilbakefall av AT/RT eller resistent sykdom.
Germinalcellesvulster i CNS
Germinalcelletumores (GCT) er en gruppe sjeldne svulster med antatt utgangspunkt i totipotente primordiale germinalceller, som migrerer til «feil plass» og kan undergå malign transformasjon i løpet av vekst og differensiering. De er heterogene både mht primærlokalisasjon, histologi, biologisk profil og behandlingsrespons. Totalt utgjør GCT ca. 3,4 % av maligne svulster hos barn, og ca. 1/5 av dem oppstår i sentralnervesystemet. Insidenskurven er to‑puklet, høyest hos sped-/småbarn med en ny stigning rundt puberteten, og de er hyppigere hos gutter
Lokalisasjon
De finnes oftest i midtlinjen, selv om andre lokalisasjoner forekommer. Særlig vanlig er pinealregionen. Undergruppen «germinomer» kan være bifokale (pinealregion + suprasellært) uten at dette regnes som metastatisk sykdom. Vanligste spredningsform er via utsæd i cerebrospinalvæsken.
Symptomer/tegn
Avhenger av lokalisasjon. Ved beliggenhet i pinealregionen oppstår ofte hydrocephalus med trykksymptomer pga aqueductstenose. Videre sees ofte øyepareser og diplopi, samt «Parinauds syndrom» (blikkparese oppad og dissossiert pupillerefleks med intakt akkomodasjon, men manglende lysrefleks). Ved suprasellær lokalisasjon omfatter symptombildet foruten synsforstyrrelser også ofte diabetes insipidus og andre endokrine dysfunksjoner, som f.eks. forsinket pubertet.
Diagnose
Ved mistanke om intracerebral GCT bør pasienten utredes videre ved spesialavdeling. MR-undersøkelsen må omfatte hele CNS-aksen i henhold til spesifiserte retningslinjer i SIOP CNS GCT II-protokollen. Spinalvæsken skal undersøkes cytologisk for utsæd av tumorceller. Det må gjøres før tumor er manipulert operativt. Dersom det har vært behov for ø.hj.-operasjon pga høyt intracranielt trykk, bør det gå minst 10 dager før undersøkelsen foretas. Tumormarkørene AFP og Beta-HCG skal analyseres i både serum og cerebrospinalvæske. Dersom disse er forhøyet (i én eller begge komponenter), kan man fastslå at det dreier seg om en «secreting germ cell tumor» i undergruppen malignt non-germinom (NG-GCT). Dermed kan biopsi unngås. Men dersom markørene er under de definerte grenseverdiene kreves histologisk verifikasjon, dog med unntak av bifocal sykdom med karakteristisk lokalisasjon, som er typisk for germinom (se ovenfor).
Utredningen må omfatte så vel utbredelsesgrad som histologisk subtype, som skissert i følgende figurerer fra SIOP-CNS GCT II-protokollen.
Rekkefølgen av undersøkelsene avhenger av om det foreligger behandlingstrengende akutte symptomer:
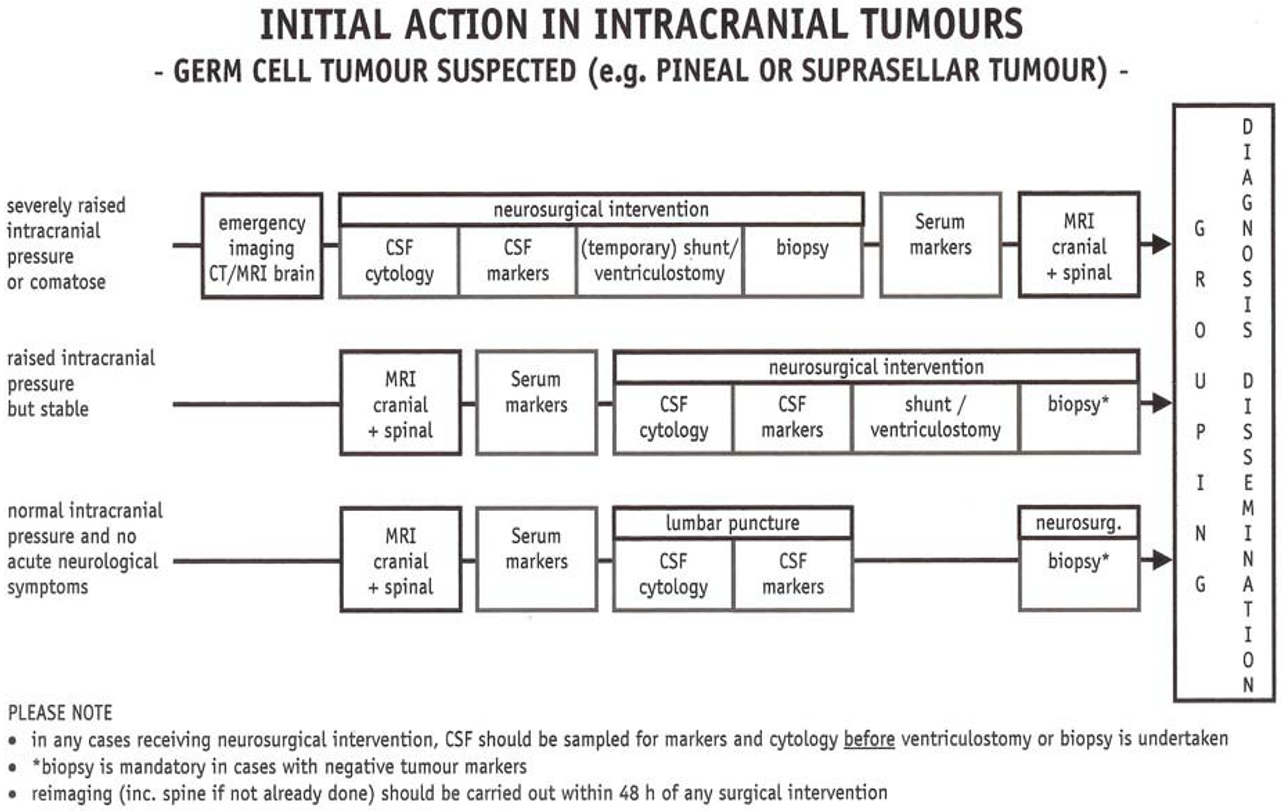
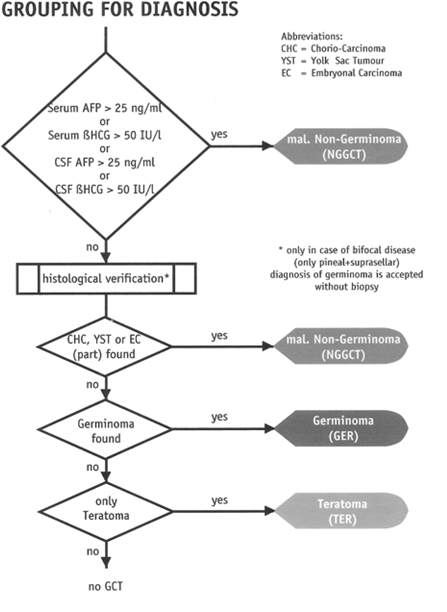
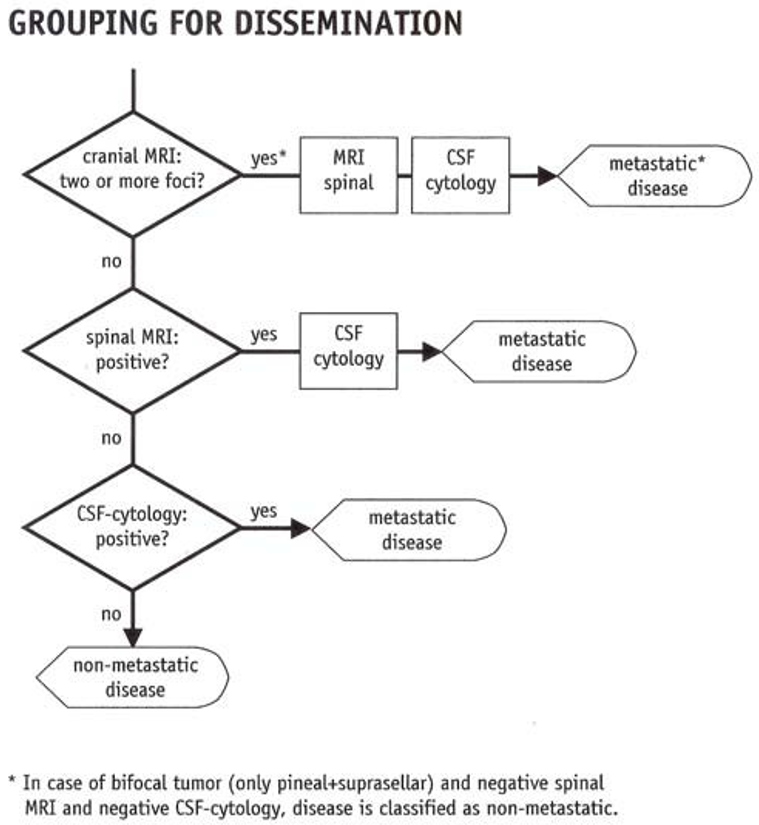
Patologi/Histologi
Germinalcellesvulster kan fremvise:
- Germinalcelle differensiering (→germinom, som tilsvarer seminom og dysgerminom i testis)
- Somatisk diffensiering:
- Embryonal differensiering (→ embryonalt carcinom)
- Ekstraembryonal (→choriocarcinom og → plommesekktumor = yolksac tumor = YST)
- Teratom (moden/umoden) med tilstedeværelse av modne eller umodne strukturer fra alle 3 germinal-lag (graderes 1–3 avhengig av relativ mengde umodent vev).
NB: Ofte forekommer blandingsformer!
Histologien hos nyfødte er nesten alltid teratom (modne og umodne), i blant med små foci av plommesekktumor. I spedbarns- og småbarnsalder dominerer YST, mens andel germinomer stiger i senere barne- og ungdomsår.
Tumormarkørene har tendens til overlapping mellom undergruppene, teratomceller kan f.eks. være noe positive for AFP. Det er avgjørende viktig for valg av behandling å undersøke for et bredt immunhistokjemisk/molekylærbiologisk/cytogenetisk panel.
Histologi | Klinikk | Tumormarkører | Følsom for | ||
|---|---|---|---|---|---|
|
| AFP | Tot. HCG | Cytostatika | RT |
Germinom | malign | – | (+) | +++ | +++ |
Malignt non-germinom (secreting GCT): |
|
|
|
|
|
Embryonalt carcinom | malign | – | – | +++ | ++ |
Plommesekktumor | malign | +++ | – | +++ | ++ |
Choriocarcinom | malign | – | +++ | +++ | ++ |
Teratom | benign (potensielt malign) | – | – | –/? | +/– |
Behandling
Både utredning og behandling av GCTs krever nevroonkologiske spesialistteam, og pasienten må snarest mulig henvises til et universitetssykehus.
Prinsippet for valg av terapi ved sammensatte GCTs er at man skal behandle i henhold til den mest maligne komponenten. Dersom full utredning mht markører og utsæd ikke er foretatt, må sykdommen behandles som metastatisk sykdom av den mest maligne undergruppe.
Det er meget viktig å sikre en fullverdig utredning nært opptil oppstart av behandling (min. 14 dager før, ellers kreves ny utredning).
Protokollen SIOP CNS GCT II er stengt for inkludering av nye pasienter, men brukes fortsatt som utgangspunkt for behandlingen av maligne germinalcellesvulster. En ny protokoll er under utarbeidelse, men avhenger av resultatene av nåværende protokoll (analyseres etter follow up avsluttes i 2020).
Generelt avhenger behandlingsintensiviteten av histologi, utbredelsesgrad, AFP-nivå og alder (grense 6 år). Strategien må korrigeres avhengig av evalueringer underveis, bl.a. med henblikk på behandlingsrespons.
Nevrokirurgi er viktig for biopsi der dette er påkrevet, samt ved behov for trykkavløsende ventriculostomi. Ved rene teratomer er kirurgi hovedbehandlingen. Kirurgisk reseksjon kan også være aktuelt ved resttumor etter kjemoterapi for andre histologiske undergrupper.
Teratom behandles i hovedsak med kirurgi. Evt. tilleggsbehandling individualiseres etter histologi, reseksjonsstatus mv. Studiesentret kan kontaktes for råd.
Lokalisert germinom behandles med kjemoterapi og ventrikulær strålebehandling med fokal boost, uansett om de er i komplett eller partiell remisjon etter kjemoterapi (ihht partiell remisjon etter kjemoterapi i protokollen (standard behandling)).
Germinom med metastaser behandles med strålebehandling mot total-CNS (CSI) ihht SIOP CNS GCT II-protokollen.
Lokalisert NG-GCT behandles med kjemoterapi og strålebehandling. Kjemoterapien er PEI-kurer (Cisplatin, Etoposid og Ifosfamid) ihht standard risiko pasienter i protokollen. For høy-risiko NG-GCT (AFP>1000 ng/ml el. diagnosealder < 6år) diskuteres valg av kjemoterapi. Studiesentret kan kontaktes for råd. Strålebehandlingen er lokal ihht SIOP CNS GCT II-protokollen, men inkludering av ventriklene vurderes, spesielt gjelder dette ved protonbestråling.
NG-GCT med metastaser behandles med kjemoterapi og strålebehandling. Kjemoterapien er PEI-kurer (Cisplatin, Etoposid og Ifosfamid) ihht standard risiko pasienter i protokollen. For høy-risiko NG-GCT (AFP>1000 ng/ml el. diagnosealder < 6år) diskuteres valg av kjemoterapi. Studiesentret kan kontaktes for råd. Strålebehandlingen mot total-CNS (CSI) ihht SIOP CNS GCT II-protokollen.
Prognose
Best prognose har germinom, med 90–95 % 5-års EFS i foregående SIOP-protokoll (CNS GCT-96) som inneværende bygger videre på. Ved NG-GCT er EFS rundt 70 %.
Oppfølging
Detaljert oppfølgningsplan både m.h.t residiv og senskader foreligger i protokollen. Residiv ved maligne GCTs er hyppigst de første 2 år etter behandling. Alle pasienter skal følges opp (inkl. klinikk, MR og markører) hver 3. måned første år og videre i h.h.t. protokoll minst 5 år etter avsluttet behandling. Ved germinom (som kan residivere enda senere) anbefales oppfølgning minst 10 år.
Barn med germinalcelletumor vil i varierende grad få senfølger av behandlingen som må følges nøye. Nevropsykologisk testing gjennomføres 1, 2 og 5 år etter diagnose i hht nasjonalt tverrfaglig oppfølgingprogram for barn med tumor cerebri, og det gjøres også en vurdering av behov for dette tidligere i forløpet. Pedagogisk oppfølging ved behov er vesentlig. I varierende grad får barnet oppfølging av barnenevrolog, fysioterapeut, endokrinolog, nyrefunksjon, hørsel, ØNH-status og øyestatus. Det er en målsetting at der bivirkninger av behandlingen påvises, etableres samarbeid med lokale aktører for å legge til rette best mulig habilitering.
For mer informasjon, se referanselisten til «SIOP CNS GCT II protocol for the diagnosis and treatment of children, adolescents and young adults with intracranial Germ Cell Tumours» (Prospective Trial for the Diagnosis and Treatment of Intracranial Germ Cell Tumors (SIOPCNSGCTII), 2011-), hvor erfaringer fra en lang rekke europeiske, amerikanske og japanske studier er omtalt (Fullstendig protokoll tilgjengelig på hjemmesidene til KSSB).
Kraniofaryngeom
Kraniofaryngeom er en histologisk benign epitelial svulst utgående fra embryonale rester av ductus craniopharyngeus.
På tross av benign histologi fører slike svulster hos mange pasienter til betydelige senskader i form av hypopituitarisme, massiv overvekt, synsforstyrrelser og kognitive skader. Dette skyldes beliggenheten i hypothalamus og nærhet til synsveiene.
Kraniofaryngeomer klassifiseres til WHO grad I.
Etiologien er ukjent og det foreligger ingen kjente genetiske disposisjoner.
Forekomst
Kraniofaryngeomene utgjør ca. 3 % av intrakranielle svulster hos barn, dvs 1–2 nye tilfeller i Norge pr år. Ingen kjønnsforskjell.
Det er to ulike manifestasjoner ved vevsundersøkelse, adamantinomatøs type som er helt dominerende hos barn, mens voksne oftest har papillær type.
Molekylærbiologi
Det er nylig påvist at barnekraniofaryngeom, den adamantinomatøse typen, har en mutasjon i genet til beta-catenin som er viktig for wnt-signalveien. Mutasjonen fører til overaktivering av beta-catenin og derved wnt. Dette aktiverer også andre signalveier «downstream» i påvirkningsrekken med tumorutvikling.
Lokalisasjon
Vanligst suprasellært, evt med intrasellær komponent. Kan være rent intrasellær, mens enkelte har svær, gjerne cystisk utbredelse, også til 3. ventrikkel. Ektopisk lokalisasjon (ekstradural) forekommer.
Symptomer
Synsaffeksjon som uttrykk for kompresjon på synsbanekrysningen og synsbaner. Vekst i hypothalamus og hypofyse gir hormonelle forstyrrelser, herunder ikke sjelden vekstforstyrrelser.
Noen debuterer med diabetes insipidus. Hypothalamuslesjon kan gi kraftig overvekt og også kognitiv svikt. Ved store svulster økt intracranielt trykk evt med hydrocephalus.
Diagnose og utredning
MR undersøkelser gir vanligvis avklarende diagnostikk. CT kan vise kalk. Ved stor cyste tappes vanligvis fra denne med funn av «maskinoljelignende» væske. Biopsi gir sikker diagnose.
Hormonutredning av hypothalamus-hypofyseaksen og diabetes insipidus er viktig. Likeledes synsundersøking, vekstforhold og kognitiv status.
Behandling
Internasjonalt har man ikke kommet frem til en klar og entydig algoritme for behandling av denne vanskelig tumortypen (Muller, 2010). Hvert enkelt barn må vurderes for et individualisert behandlingstilbud basert på vurderinger ut fra pasientens alder, kliniske tilstand og tumors størrelse og vekstmønster.
Kirurgi har tradisjonelt vært primær behandling hvor man har tilstrebet radikalitet. Det er imidlertid en økende internasjonal trend å være skånsom ved reseksjon for å unngå skade på hypothalamus (Puget et al., 2007). Hypothalamuslesjon er en følge av tumors infiltrasjon, men det har vist seg at ved kirurgi i denne regionen kan sekvelene øke betydelig. En tilstreber derfor kirurgi av tumordelen utenfor hypothalamus, ikke minst hvis det er påvirkning av synsveiene. Biopsi gjøres ved diagnose.
Ved inkomplett reseksjon kan postoperativ strålebehandling / stråleknivbehandling være aktuelt for å redusere risiko for videre vekst. En større tysk studie viser at en ikke taper noe ved å utsette en slik behandling inntil det har inntruffet vekst av tumor etter kirurgien.
Intracystisk behandling. Hos barn er de fleste svulstene cystiske og ved store cyster kan man initialt implantere kateter for tømming av cystene og avlasting av press på omgivelsene. Instillasjon i cystene av cellegift (bleomycin) eller isotop (Yttrium) har tidligere vært brukt i behandlingen. Komplikasjonene har imidlertid vært utfordrende og behandlingen komplisert. I de senere år har instillasjon av interferon blitt standardbehandling ved flere sentra. Man oppnår ikke radikal fjerning av tumor, men avlastning av synsnerver og omkringliggende vev med stor grad av symptomfrihet. Pasienten må følges med regelmessige billedoppdateringer til nydanning av cystene opphører og hvis det ikke er mulig å kontrollere cystene med tapping/ interferon kan det være indisert med annen kirurgisk behandling evt i kombinasjon med stråleterapi.
Systemisk medikamentell behandling er i dag på forsøksstadiet. Signalveier «downstream» for wnt-signalveien aktiveres ved kraniofaryngeom, for eksempel MAPK-signalveien (RAS/RAF/MEK/ERK). En har derfor nå i England startet forsøk med MEK-inhibitor i utvalgte tilfeller. Resultater foreligger ikke nå.
En annen tilnærming er antiinflammatorisk behandling, da inflammasjon er en viktig del av patogenesen ved disse svulstene. Aktuelt er Interleukin-1 Reseptor-inhibitor (anakinra). Kliniske forsøk vil komme.
Symptomatisk behandling: Det er vesentlig at barn med slike svulster følges opp i et multidisiplinært team (MDT) med bred erfaring innen nevrokirurgi, nevroonkologi og nevroendokrinologi. Hormonutfall må monitoreres og substitueres. Vektøkning må prøves begrenset Videre er det viktig med oppfølging av kognitive funksjoner, synsfunksjon osv.
Prognose
Selv etter radiologisk tilsynelatende radikal kirurgisk reseksjon ser man opp til 15 % residiv. Pasientene som er behandlet med intracystisk interferon har svært gode resultater hva angår symptomkontroll, men oppfølging blir som ved en kronisk sykdom. Mortaliteten ved kraniofaryngeom er svært lav, men som fremgår er det høy morbiditet.
Oppfølging
Oppfølging avhenger av valgt behandlingsregime. Ved både cystebehandling og tradisjonell kirurgisk behandling er det tett oppfølgning, da residiv forkommer hyppig de første 5 år. Det er risiko for sent residiv, så kontroller (klinisk og MR), bør gjennomføres med lengre intervall gjennom flere tiår.
Nøye symptomatisk oppfølging er helt vesentlig.
Barn/ungdom med kraniofaryngeom kan i varierende grad få senfølger av behandlingen som må følges. Nevropsykologisk testing gjennomføres 1, 2 og 5 år etter diagnose i hht nasjonalt tverrfaglig oppfølgingprofgram for barn med tumor cerebri, og det gjøres en vurdering av behov for dette også tidligere i forløpet. Pedagogisk oppfølging ved behov er vesentlig. Det er en målsetting at der bivirkninger av behandlingen påvises, etableres samarbeid med lokale aktører for å legge til rette best mulig habilitering når det er behov for dette.
Plexus chorioideus-svulster
Epidemiologi
Plexus choroideus svulster utgjør kun 2–4 % av hjernesvulstene hos barn. De fleste pasientene er mellom 0–3 år på diagnosetidspunktet.
Symptomer
Små barn har som oftest en raskt økende hodeomkretsutvikling, eldre barn utvikler generelle trykksymptomer.
Billedundersøkelse
Svulsten er utgående fra plexus choroideus, vevet som produserer CSF i hjernes naturlige hulrom. Svulster i plexus choroideus gir overproduksjon av CSF og ledsagende hydrocephalus er et typisk funn. Hos små barn ser man svulsten i en av sideventriklene eller tredje ventrikkel. Hele CNS-aksen må avbildes for eventuelle metastaser (carcinomer).
Patologi/ Klassifisering
WHO klassifiseringen skiller mellom 3 ulike histologiske grader: Plexus choroideus papillom (WHO grad I), atypisk plexus choroideus papillom (WHO grad II) og plexus choroideus carcinom (WHO grad III).
Diagnosen stilles ved histologisk undersøkelse.
Prognosen
Plexus choroideus papillomer har ved total kirurgisk fjerning en svært god prognose (kurativt).
Plexus choroideus carcinomer har i de fleste rapporter en 5 års overlevelse på under 50 %.
Behandling
Kirurgi er grunnlaget for behandling av plexussvulster hvor målet er total fjerning av tumor. De fleste pasientene er på behandlingstidspunktet i første leveår og har et lite blodvolum, svulstene har stor blodgjennomstrømning, som oftest med en betydelig hydrocefalus. Det kirurgiske inngrepet er derfor utfordrende. Mange vil kreve oppfølgning og eventuell behandling for hydrocephalus i forløpet.
Pasienter med plexus papillom (WHO grad I og II) skal ikke ha tilleggsbehandling, men skal følges opp over tid (regelmessige MR undersøkelser)
Plexus choroideus carcinom (WHO grad III) er en aggressiv tumor. Prognosen er noe bedre ved total fjerning av tumor. Mange har sykdom med spredning og eller innvekst sentralt i hjernen allerede på diagnosetidspunktet og vil være avhengig av tilleggsbehandling.
Plexus choroideus carcinomene er følsomme for kjemoterapi med kombinasjoner av carboplatin, etoposid, vincristin og cyklofosfamid. Stråleterapi forbedrer prognosen, men pasientens alder vil ofte være en begrensende faktor (de fleste vil være for unge). Det er ingen konsensus omkring optimal behandling.
Oppfølging: Alle barn kontrolleres med tanke på tumorkontroll etter 3, 6, 12 18 og 24 mnd med MR, deretter MR minimum to ganger årlig fra 2 til 5 år etter avsluttet behandling, og senere årlig.
Barn med plexuscarcinomer vil i varierende grad utvikle senfølger av behandlingen som må følges nøye. Nevropsykologisk testing gjennomføres i hht nasjonalt tverrfaglig oppfølgingsprogram for barn med hjernesvulst 1, 2 og 5 år etter diagnose, og det gjøres en vurdering av behov for dette også tidligere i forløpet. Pedagogisk oppfølging ved behov er vesentlig.
Det anbefales oppfølging av barnenevrolog, fysioterapeut, endokrinolog, nyrefunksjon, hørsel, ØNH-status og øyestatus. Der bivirkninger av behandlingen påvises, etableres det et samarbeid med lokale aktører for å legge til rette best mulig habilitering.
Solide svulster utenfor sentralnervesystemet
Sist faglig oppdatert: 26.05.2020
Sarkomer
Sarkomer er maligne svulster som utgår fra mesenkymalt vev. Det er den nest vanligste formen for solid, malign svulst utenfor sentralnervesystemet i barnealder (Childhood cancer in the Nordic countries, 2008).
Sarkomer inndeles i bensarkomer og bløtvevsarkomer og får navn etter det vevet de har fellestrekk med.
Bensarkomer inndeles i osteogent sarkom (Kager et al., 2003) og Ewing sarkom, sistnevnte kan også opptre i bløtvev (Principles and practice of pediatric oncology, 2006).
Bløtvevsarkomer inndeles i rabdomyosarkom som utgår fra celler som er eller skal bli muskelceller, og non-rabdomyosarkom. Non-rabdomyosarkom er en heterogen gruppe tumores som består av mer enn 30 ulike undergrupper (Principles and practice of pediatric oncology, 2006).
Se også «Nasjonalt handlingsprogram med retningslinjer for utredning, behandling og oppfølging av sarkom» (Helsedirektoratet, 2018).
Epidemiologi
Insidensen av sarkom hos barn er 1,49 pr 100 000, det vil si at 9,5 % av barna som får en kreftdiagnose, har sarkom. Bløtvevsarkom er hyppigst og forekommer hos 6 % av de kreftsyke barna. Bensarkom er sjeldnere og utgjør 3,5 % av krefttilfellene hos barn under 15 år (Childhood cancer in the Nordic countries, 2008).
Symptomer og funn
Osteogent sarkom debuterer oftest med smerter. I begynnelsen er smertene ofte intermitterende, men etter hvert blir de konstante og mer intense. Tumorrelatert hevelse og nedsatt funksjon i naboledd oppstår mye senere. I ca 10 % av tilfellene debuterer osteogent sarkom som patologisk fraktur (Kager et al., 2003).
Ewing sarkom debuterer oftest med smerter. Smertene er vedvarende og kan oppstå uavhengig av aktivitet, for eksempel i hvile eller om natten. Eventuelle tilleggssymptomer avhenger av tumors lokalisasjon og størrelse. Subfebrilitet er rapportert hos ca 1/3 av pasientene, først og fremst hos de med metastatisk sykdom på diagnosetidspunktet (Paulussen, Kovar, & Jürgens, 2005).
Hvordan et bløtvevsarkom debuterer, avhenger av tumors lokalisasjon og størrelse. Hvis ikke andre strukturer komprimeres, gir sarkom sjelden uttalte symptomer og oppdages ofte tilfeldig grunnet tumors størrelse.
Bløtvevsarkom kan gi smerter hvis de presser på andre strukturer eller obstruerer avløp som for eksempel i blæreregionen. Vedvarende nesetetthet og sekresjon fra nesen er vanlige symptomer ved sarkom i nasofarynks. Vaginale polypper og utflod er vanlig ved sarkom i uterus og vagina (Bisogno & Bergeron, 2005).
Allmenn sykdomsfølelse og vekttap forekommer svært sjelden i startfasen ved sarkom, men forekommer hyppigere ved avansert sykdom.
Utredning
Utredning av sarkom hos barn og ungdom bør skje ved Barne-/ungdomsavdelinger med spesialkompetanse i håndtering av sarkom eller et sarkomsenter. Det er svært viktig at utredningen skjer i henhold til gjeldende protokoller. Man bør mistenke sarkom ved tumor i benvev eller tumor beliggende dypt i bløtvev.
Utredning med tanke på diagnostikk og stadieinndeling gjøres i henhold til aktuelle behandlingsprotokoller og må inneholde adekvat billeddiagnostikk av primærtumor og nøyaktig evaluering av potensielle metastaser.
Radiologiske undersøkelser
Ved mistanke om osteogent sarkom er det viktig med vanlig røntgenundersøkelse av det aktuelle området, gjerne supplert med CT for å se på skjelettstruktur, og MR for å evaluere benmargs- og bløtdelsaffeksjon samt tumors relasjon til årer og nerver. Søk etter metastaser konsentreres om skjelett og lunger for >95 % av metastasene ved osteogent sarkom opptrer i disse lokalisasjonene. Røntgen og CT av lungene benyttes for å kartlegge eventuelle lungemetastaser. MR av affisert knokkel skal utføres for å lete etter skip-metastaser. MR og NaF-PET bør utføres for å kartlegge eventuelle skjelettmetastaser. FDG-PET/CT er en nyttig tilleggsundersøkelse for kartlegging av sykdomsutbredelse ved osteogent sarkom.
Ved utredning av Ewing sarkom benyttes vanlig røntgen, MR og CT for å kartlegge sykdoms-utbredelse på primærtumorstedet. CT egner seg best til å vise endringer i korteks- og skjelettstruktur, mens MR egner seg best for kartlegging av tumorutbredelse i benmarg og bløtvev samt for å evaluere tumors relasjon til nærliggende strukturer som årer og nerver. Det er viktig å dokumentere tumors utbredelse i 3 dimensjoner. CT thorax anbefales for kartlegging av eventuelle lungemetastaser. MR anbefales for kartlegging av eventuelle skjelettmetastaser. FDG-PET/CT er en nyttig tilleggsundersøkelse for kartlegging av sykdomsutbredelse ved Ewing sarkom.
Ved mistanke om bløtvevsarkom bør man benytte MR, ultralyd og eventuelt CT av primærtumor-stedet. Det er viktig å kunne angi tumors størrelse i 3 dimensjoner ved diagnose, slik at man kan evaluere behandlingsrespons ved senere evalueringstidspunkter. MR er å foretrekke for de fleste lokalisasjoner og er obligatorisk ved primærtumor i genitourinaltraktus og ved paraspinal tumor. CT kan være et supplement ved spørsmål om subtile bendestruksjoner. Røntgen thorax og CT thorax benyttes for å oppdage lungemetastaser. Ultralyd abdomen benyttes for å kartlegge lymfeglandelaffeksjon og levermetastaser. FDG-PET/CT er en nyttig tilleggsundersøkelse for kartlegging av sykdomsutbredelse ved bløtvevsarkom.
Biopsi
Sarkomdiagnosen må alltid bekreftes histologisk. Målet med biopsitaking er å få nok tumormateriale til histologi, immunhistokjemi, cytogenetikk, sentralisert «review», samt vev i reserve for eventuelle supplerende analyser.
Biopsitaking skjer oftest ultralydveiledet, men kan også skje CT-veiledet. Det er nyttig å starte med finnålsaspirasjon og fortsette med tru-cut-biopsi hvis forholdene ligger til rette for det. Dersom denne fremgangsmåten ikke kan sikre nok materiale for å kunne stille en sikker diagnose er det aktuelt å supplere med åpen biopsi. Det er viktig å ta med i betraktning at biopsikanalen og eventuelle arr må inkluderes «en-bloc» ved etterfølgende kirurgi, så det er viktig at affeksjon av nevrovaskulære strukturer i størst mulig grad unngås. På ekstremiteter bør incisjoner legges longitudinelt. Farging f.eks. med metylenblått bør benyttes for markering av biopsikanal. Endoskopisk biopsitagning er akseptabelt ved tumor i blære, prostata og vagina.
Benmargsaspirasjon og eventuelt benmargsbiopsi hører med i utredningen av enkelte typer bløtvevsarkom.
Spinalpunksjon for å sjekke om det foreligger CNS-affeksjon, er kun nødvendig ved CNS-nær tumor.
Behandling
Sarkom i barnealder utredes og behandles i henhold til internasjonale protokoller. For øyeblikket behandles osteogent sarkom etter anbefalinger fra den avsluttede EURAMOS 1-protokollen, Ewing sarkom behandles i henhold til EuroEwing 2012-protokollen, mens residiv av Ewing sarkom og refraktært Ewing sarkom behandles i henhold til rEECur-protokollen. Rabdomyosarkom behandles i henhold til anbefalinger fra den avsluttede EpSSG RMS 2005-protokollen og høygradig malignt non-rabdomyosarkom behandles i henhold til anbefalinger fra den avsluttede EpSSG NRSTS 2005 (https://www.skion.nl/). Når det gjelder rabdomyosarkom, har fagmiljøet i Norge søkt REK og SLV om å få godkjent at norske pasienter kan inkluderes i og behandles i henhold til den nye internasjonale behandlingsprotokollen fra EpSSG, nemlig «FaR-RMS: An overarching study for children and adults with Frontline and Relapsed RhabdoMyoSarcoma». Forhåpentligvis kan norske pasienter få tilbud om være med i denne studien allerede i løpet av 2020. Studien er i regi av EpSSG og behandlingen er basert på resultatene fra RMS-2005-protokollen. Behandlingen av sarkom i barnealder er i hovedtrekk multimodal og består av kjemoterapi, kirurgi og/eller bestråling.
Medikamentell behandling
Behandlingen av osteogent sarkom består av pre- og postoperativ kjemoterapi samt kirurgi. Kjemoterapien baserer seg på høye doser metotreksat, cisplatin og doksorubicin (Bielack et al., 2015; Smeland et al., 2019)
Ewing sarkom er generelt en kjemosensitiv tumorform og induksjonsbehandlingen består per i dag av vinkristin, doksorubicin, cyklofosfamid, ifosfamid og etoposid (EE2012-protokollen: https://birmingham.ac.uk/research/activity/mds/trials/crctu/trials/EE2012/index.aspx). Ved dårlig histologisk eller radiologisk respons, ved stor primærtumor (>200 ml) eller ved metastatisk sykdom kan man vurdere å intensivere kjemoterapien med høydosebehandling med autolog stamcellestøtte (https://www.skion.nl/workspace/uploads/Protocol-EpSSG-RMS-2005-1-3-May-2012_1.pdf). Ved behandlingsrefraktær sykdom og residiv består behandlingen av intensiv kjemoterapi og det er flere ulike kjemoterapiformer som har vist seg å ha effekt:
- cyklofosfamid/topotekan,
- irinotekan/temozolamid,
- gemcitabin/docetaxel og
- høydose ifosfamid.
I rEECur-studien randomiseres pasientene mellom de ulike behandlingsarmene for å finne ut hvilken av de fire armene som fungerer best. Foreløpige resultater fra rEECur-studien tyder på at gemcitabin/docetaxel har dårlig effekt og denne behandlingen anbefales derfor ikke.
Rabdomyosarkom er som regel en kjemosensitiv tumorform, og kjemoterapi benyttes derfor i behandlingen hvis man ikke kommer til målet med kirurgi alene. Kjemoterapi benyttes for å redusere tumorstørrelsen slik at radikal kirurgi, evt annen lokalbehandling, kan bli mulig. Aktuell kjemoterapi består av ifosfamid, vinkristin, actinomycin D og eventuelt doxorubicin. Etter avsluttet primærbehandling kan det være aktuelt med vedlikeholdsbehandling med cyklofosfamid og vinorelbin i lave doser (https://www.skion.nl/workspace/uploads/Protocol-EpSSG-RMS-2005-1-3-May-2012_1.pdf). I den nye behandlingsprotokollen (FaR-RMS) vil dessuten irinotekan, temodal og regorafenib inngå i behandlingen av enkelte pasientgrupper.
Non-rabdomyosarkom er en svært heterogen gruppe sarkomer, og de fleste er ikke like kjemo-sensitive som rabdomyosarkom. Den europeiske studien EpSSG NRSTS 2005 (skion.nl/workspace/uploads/EpSSG-NRSTS-version-1-1.pdf) inndeler non-rabdomyosarkom i 3 ulike hovedgrupper; synovialt sarkom, «voksen type» bløtvevsarkom og andre typer bløtvevsarkom. Synovialt sarkom er desidert den vanligste formen for non-rabdomyosarkom i barnealder. Denne sarkomtypen er relativt kjemosensitiv, og hvis det ikke ligger til rette for radikal kirurgi i utgangspunktet, anbefales det kjemoterapi med ifosfamid/doxorubicin for å redusere tumorstørrelsen før eventuell kirurgi. Ved «voksen type» bløtvevsarkom er effekten av kjemoterapi mer usikker, men EpSSG NRSTS 2005 anbefaler kjemoterapi i form av ifosfamid/doxorubicin dersom man ikke kan foreta radikal kirurgi i utgangspunktet (https://skion.nl/workspace/uploads/EpSSG-NRSTS-version-1-1.pdf).
Rabdoid tumor utenfor sentralnervesystemet er en undergruppe av non-rabdomyosarkom. Den er sjelden, men svært aggressiv og ofte letal i barnealder. Grunnet høy dødelighet av denne tumortypen er det svært viktig med adekvat behandling. EpSSG NRSTS 2005 anbefaler et intensivt kjemoterapiregime med vinkristin/doxorubicin/cyklofosfamid alternerende med ifosfamid/etoposid hver annen uke (https://skion.nl/workspace/uploads/EpSSG-NRSTS-version-1-1.pdf). Alternativt kan rabdoid tumor behandles i henhold til anbefalingene fra det multinasjonale European rhabdoid registry (EU-RHAB). EU-RHAB anbefaler et intensivt kjemoterapiregime med doksorubicin, ifosfamid, karboplatin, etoposid, vinkristin, actinomycin D og cyklofosfamid, evt supplert med høydosebehandling med karboplatin og thiotepa, etterfulgt av autolog stamcellestøtte.
Kirurgisk behandling av rabdomyosarkom (RMS)
Lokal kontroll er nødvendig for å kurere barn med RMS, og dette kan oppnås ved kirurgi og/eller strålebehandling (evt brachyterapy). Konservativ tilnærming er anbefalt, og preoperativ kjemoterapi for reduksjon av tumor er vanlig før tumorreseksjon/bestråling.
Målet med lokalbehandling er kurasjon uten eller med minst mulig senskader. Hvilken type lokalbehandling som egner seg best avhenger av primærtumors lokalisasjon og størrelse, pasientens alder og kjemoterapirespons. Kirurgisk planlegging må inkludere alle rekonstruktive prosedyrer med optimal timing av eventuell strålebehandling i tillegg (https://www.skion.nl/workspace/uploads/Protocol-EpSSG-RMS-2005-1-3-May-2012_1.pdf).
Anbefalinger:
- Sarkom i barnealder utredes og behandles i henhold til internasjonale protokoller som krever samtykke fra foreldrene.
- Osteogent sarkom behandles i henhold til anbefalinger i den avsluttede EURAMOS 1-protokollen.
- Ewing sarkom behandles primært i henhold til EuroEwing 2012-protokollen og i henhold til rEECur-protokollen ved residiv.
- Rabdomyosarkom behandles i henhold til anbefalinger i den avsluttede EpSSG RMS 2005- protokollen. I løpet av 2020 vil det være aktuelt med behandling i henhold til EpSSG-studien FaR-RMS.
- Non-rabdomyosarkom behandles i henhold til EpSSG NRSTS 2005-protokollen.
Lymfeknuter
Målet er å påvise lymfeknuteaffeksjon ved «sampling» og unngå radikalt lymfeknutetoilette. «Sentinel node mapping» med for eksempel methylenblått anbefales.
For spesifikke lokalisasjoner og detaljer forøvrig vises det til Protokoll EpSSG RMS2005, versjon 1.4 international – Dec 2013 (https://www.skion.nl/workspace/uploads/Protocol-EpSSG-RMS-2005-1-3-May-2012_1.pdf) og etter hvert også protokollen FaR-RMS
Kirurgisk behandling av non-RMS bløtvevssarkom (non-RMS STS)
Her er det tilnærmet identiske kirurgiske grunnprinsipper som for RMS. Det vises for øvrig til protokoll EpSSG NRSTS 2005 (https://skion.nl/workspace/uploads/EpSSG-NRSTS-version-1-1.pdf).
Kirurgisk behandling av bensarkomer
Prinsippet er at tumor må fjernes med fri margin. Det er viktig med nøye planlegging i forkant av inngrepet. Den kirurgiske behandlingen må passe inn i den multimodale behandlingen uten å forsinke øvrig behandling. Se «Nasjonalt handlingsprogram med retningslinjer for utredning, behandling og oppfølging av sarkom» (Helsedirektoratet, 2018).
Strålebehandling
Dersom man ikke oppnår lokal kontroll med kirurgi, kan det være aktuelt med supplerende strålebehandling. Strålebehandling påvirker vekst og utvikling og skal benyttes med varsomhet til barn og ungdom.
Osteogent sarkom er svært lite strålesensitiv, og strålebehandling inngår derfor ikke i rutinebehandlingen, men benyttes kun ved inoperabilitet og i palliativt øyemed.
Ewing sarkom er svært strålesensitiv. Strålebehandling inngår rutinemessig i behandlingen av Ewing sarkom hvis man ikke oppnår fullstendig lokal kontroll med kjemoterapi og kirurgi, det vil si ved marginal og intralesjonell reseksjon, samt ved inoperabilitet. Strålebehandling benyttes dessuten i behandlingen av lunge- og skjelettmetastaser ved Ewing sarkom.
Rabdomyosarkom er strålesensitiv, og strålebehandling inngår som ledd i behandlingen hvis ikke histologien er lite aggressiv og lokalisasjonen gunstig, eventuelt at kirurgien er komplett. Strålebehandling av rabdomyosarkom gjennomføres fortrinnsvis parallelt med postoperativ kjemoterapi.
I gruppen non-rabdomyosarkom er strålefølsomheten variabel, og det er kun enkelte entiteter som synovialt sarkom, «voksen form» for bløtvevsarkom og rabdoid tumor utenfor sentralnervesystemet som behandles med strålebehandling. Både primærtumor og lymfeglandelmetastaser behandles. Strålebehandling gis fortrinnsvis parallelt med postoperativ kjemoterapi (https://skion.nl/workspace/uploads/EpSSG-NRSTS-version-1-1.pdf).
Oppfølging og etterkontroll
Etter avsluttet behandling for sarkom er det viktig å utføre en sluttevaluering før man starter med regelmessige kontroller. Dette for å evaluere effekten av behandlingen og for å ha et utgangspunkt for senere kontroller. Sluttevalueringen foretas vanligvis 4–6 uker etter avsluttet behandling og omfatter alltid generell klinisk undersøkelse, billeddiagnostikk av affiserte tumorområder, røntgen thorax og blodprøver. Ytterligere detaljer er beskrevet i hver enkelt protokoll. Etterkontrollene skjer vanligvis hver 3. måned de første 2–3 årene, deretter sjeldnere. Hensikten med kontrollene er både å oppdage et eventuelt residiv så tidlig som mulig og å kartlegge eventuelle seneffekter av behandlingen, både på kort og lang sikt. Alle som er behandlet for sarkom, bør følge vanlige kontroller med henblikk på: Høyde og vekt, blodtrykk, Tanner stadium, testikkelvolum, tidspunkt for menarke, skoleprestasjoner, hjertefunksjon, hørsel, nyrefunksjon og eventuelle atferdsproblemer. Detaljer for oppfølging er beskrevet i hver enkelt protokoll.
Se ellers kapittelet om senskader.
Behandling av metastatisk sykdom
Hos enkelte barn er sarkom metastatisk på diagnosetidspunktet. Behandlingen av metastatisk sykdom hos barn baserer seg på de samme prinsipper som ikke-metastatisk sykdom, men kjemoterapien er gjerne enda mer intensiv.
Livsforlengende og palliativ behandling
Ved behov for palliativ behandling hos barn har man ikke lenger et kurativt siktemål, og hensikten med behandlingen vil være å forsøke å lindre plagene så mye som mulig. Dette kan skje ved lavdosekjemoterapi og strålebehandling for å forsinke tumorvekst og lindre smerter, eventuelt smertelindrende medikamenter. Hjemmebehandling og et ordinært liv tilstrebes så lenge som mulig. Viser for øvrig til nasjonale retningslinjer for palliativ behandling hos barn (Helsedirektoratet, 2017).
Nyresvulster
Nefroblastom er den vanligste nyresvulsten i barnealder og kalles til daglig oftest Wilms tumor etter Max Wilms som beskrev den i 1899.
Dette er en ondartet svulst med utgangspunkt i embryonale nyreceller og utgjør ca 90 % av ondartede nyresvulster i barnealder. Opptrer oftest som en enkelt svulst i en nyre, men det kan være multiple svulster, og de kan også opptre i begge nyrer (bilateral svulst). Svulsten diagnostiseres oftest i småbarnsalder og er sjelden hos større barn og ungdommer. Median alder ved diagnose 3,5 år. Over 80 % er under 5 år. Kan også forekomme opp i voksen alder.
Nefroblastom rammer ca 1 av 10 000 barn. Det er ca 6–8 nye tilfeller med nefroblastom i Norge pr år.
Overlevelsen ved nefroblastom er nå gjennomsnittlig ca. 90 % (Pritchard-Jones & Pritchard, 2004).
Genetikk
Nefroblastom forkommer enten sporadisk, i assosiasjon med visse syndromer (se senere) og hos 1–2 % av pasientene som familiær sykdom. Ved familiær sykdom synes genetikken å være kompleks med flere mulige gener involvert.
Kartlegging av genetikk og molekylærbiologi ved nefroblastom er i stadig utvikling. Og man venter at nyere molekylærbiologisk forskning vil kunne identifisere genetiske endringer av betydning for risikoinndeling, behandling og prognose både ved sporadiske og arvelige former. Det vises også til kapittelet Genetikk.
Nefroblastomatose: Betegner en tilstand hvor det i den tidlige nyreutviklingen har blitt værende igjen umodne små foci av embryonale celler (nefrogenic rests) i nyreparenchymet (Beckwith, 1998). Disse vil kunne utvikle seg videre til større hyperplastiske knuter og evt til nefroblastomsvulster. Disse oppdages enten ved at de er så store (hyperplastiske) at de sees som knuter i nyrene på ultralyd ved utredning av nyretumor, eller som tilfeldig funn. De kan også være helt mikroskopiske og finnes ved undersøkelse av gjenværende nyrevev i nyre fjernet grunnet tumor. Det vil da være økt risiko for ny tumor i gjenværende, kontralaterale nyre.
Diagnose
I nesten 50 % av tilfellene har barna ingen symptomer, og svulsten oppdages da tilfeldig enten i forbindelse med andre undersøkelser i helsetjenesten eller ved at foreldre eller andre kjenner at det er en kul i barnets mage. Vel 50 % vil ha magesmerter, få stor, utspilt mage eller ha blod i urinen. Og ca 5 % vil ha tydelig nedsatt allmenntilstand.
Høyt blodtrykk, trombocytose eller hevelse i pungen kan være ledsagende symptomer.
Diagnostikken består av full klinisk undersøkelse hvor man spesielt må være forsiktig ved palpasjon av abdomen, og unngå å trykke hardt på svulsten som kan være skjør. Det er ingen spesifikk tumormarkør knyttet til nefroblastom. Diagnosen stilles ved bildediagnostikk, ultralyd og MR. Fordi 90 % av alle ondartede svulster i nyre er Wilms tumor er det vanlig å starte behandling med cellegift uten forutgående vevsprøve. Vevsprøve forbeholdes de tilfeller hvor diagnosen er usikker etter MR, hvor barnet er yngre enn 7 måneder eller eldre enn 16 år.
Tilstander som disponerer for nefroblastom
Nefroblastom er sterkt assosiert med enkelte syndromer/utviklingsforstyrrelser som:
- Aniridi
- Hemihypertrofi
- Genital/urinveis-midannelser
- Beckwith-Wiedemanns syndrom
- Denys-Drash syndrom
- WAGR syndrom
Pasienter med disse syndromene/utviklingsforstyrrelsene skal kartlegges og tilbys jevnlig kontroll der dette er indisert.
Klassifisering
Klassifiseringen følger den behandlingsprotokollen vi nå bruker:
Umbrella-protokoll SIOP-RTSG 2016. Studien vil åpnes i Norge i løpet av kort tid, men allerede nå brukes den som grunnlag for klassifisering og behandling. Denne protokollen har retningslinjer for behandling av alle typer nyresvulster.
Det er nasjonal enighet om å delta i dette multinasjonale samarbeid for nyresvulster hos barn i regi av SIOP. Det er etablert en organisasjon SIOP Renal Tumour Study Group (www.siop-rtsg.eu) som leder arbeidet. Norge deltar der sammen med de fleste nordiske land. SIOP har gjennomført kliniske studier for nefroblastom fra 1971. Det har vært økende tilslutning av deltagende land. Norge deltok tidligere mest i de engelske nefroblatomstudiene, og sluttet seg, på nasjonal basis, til SIOP-studiene da England også gjorde det fra 2001.
SIOP Renal tumour Study Group har også et nært samarbeid med nyretumorgruppen i COG (Children’s Oncology Group) i USA (http://www.childrensoncologygroup.org/, www.curesearch.org)
Den største forskjell i behandling mellom studiene til SIOP-RTSG og COG er at SIOP-RTSG foretrekker preoperativ kjemoterapi for om mulig å skrumpe tumor før kirurgi og gjøre den lettere håndterbar, mens COG går direkte til primær kirurgi der dette er mulig. De medikamentene som brukes er vinkristin, aktinomycin D og for enkelte grupper også doxorubicin (Kaste et al., 2008; Pritchard-Jones & Pritchard, 2004).
Vår klassifisering bygger på utbredelse etter preoperativ kjemoterapi, og endelig stadiesetting kommer først etter den utsatte kirurgien.
Ved diagnosetidspunktet deles pasientene inn i følgende kategorier:
- lokalisert sykdom
- metastatisk sykdom
- synkron bilateral sykdom
Preoperativ behandling gis ut fra dette. Diagnosen vil oftest baseres på bildediagnostikk alene. Biopsi/cytologi kan bli nødvendig ved usikre funn på MR eller dersom barnet er yngre enn 7 måneder eller eldre enn 16 år, men det er i henhold til den nyeste behandlingsprotokollen altså unødvendig i de fleste tilfeller.
Ultralyd av abdomen er obligatorisk. Metoden kan skille ut cystiske komponeneter og er nyttig for å oppdage små tumores i kontralateral nyre, tumorinnvekst i nyrevene og vena cava, samt metastaser til lever og abdomen. Man får også mål til volumberegning av svulsten ved diagnose. Samme mål gjentas preoperativt etter kjemoterapi.
Rtg.thorax front og side samt CT thorax er også obligatorisk i siste behandlingsprotokoll.
Videre suppleres bildediagnostikken med MR abdomen (alternativt CT abdomen) og evt preoperativ MR eller CT angiografi (for framstilling av blodkar og deres relasjon til tumor).
Nålebiopsi ved nyretumor hos barn skal kun gjøres ved usikkerhet rundt diagnosen etter radiologisk utredning, ved alder yngre enn 7 måneder eller eldre enn 16 år.
Biopsi med Tru-Cut nål utføres av radiolog i generell anestesi, og med patolog til stede for å sikre adekvat vevsmengde. Sykehusets retningslinjer for intervensjon må følges.
Ved bilateral sykdom eller mistanke om nefroblastomatose er det egne anbefalinger med ytterligere utsettelse av kirurgien ved forlenget preoperativ behandling – se protokoll.
Postoperativ behandling: Hele den postoperative behandlingsstratifiseringen hviler på patologens stadieinndeling og histologiske vurdering av operasjonspreparatet etter preoperativ behandling.
| Stadium 1 | Lokalisert til nyren, radikalt fjernet |
| Stadium 2 | Tumorvekst utenfor nyren, men radikalt fjernet |
| Stadium 3 | Tumorvekst utenfor nyren, ikke radikalt fjernet |
| Stadium 4 | Fjernmetastaser (oftest i lungene) |
| Stadium 5 | Tumor i begge nyrer (Bilateral sykdom). |
Histologien deles i lav-, intermediær- og høyrisiko etter bestemte kriterier.
Kombinasjonen av stadium og histologisk risikogruppe bestemmer så hvilken postoperativ kjemoterapi som skal gis, og hvorvidt det også skal suppleres med strålebehandling – se protokoll for detaljer.
Umbrella-protokoll SIOP-RTSG 2016 har som mål gjennom internasjonalt samarbeid å samle pålitelige data som kan brukes til å besvare spørsmål som har direkte klinisk relevans for pasienter. Man ønsker å samle tumorvev samt blod og urin for å lete etter nye markører for å forbedre en risikotilpasset stratifisering av behandlingsintensiteten. Protokollen vil åpnes for å inkludere norske pasienter innenfor kort tid.
Prognosen ved nefroblastom nå er så god at man vil også forsøke å designe behandlingsprogrammer som reduserer risiko for senskader (antracykliner innebærer en risiko for kardiale senskader), og opprettholder de gode behandlingsresultatene.
Andre målsettinger i studien er å forsøke å bedre resultatene ved å optimalisere seleksjonen (riktig stadie/riktig histologi) ved sentral «review» av både histologi og radiologi på alle pasienter
Langtidsoppfølging av barn med nyresvulster
Kirurgi, kjemoterapi og eventuell strålebehandling kan gi senfølger som manifesterer seg kort eller lang tid etter behandling. Nefroblastompasienter har økt risiko for hypertensjon og nedsatt nyrefunksjon. Strålebehandling vil gi senfølger av ulik grad avhengig av omfanget for strålebehandlingen (påvirkning av tarmfunksjon, skoliose, lungeforandringer ved stålebehandling av metastaser, sekundær cancer m.m). Bruk av antracykliner i behandlingen samt evt bestråling av thorax er risikofaktorer for kardiale senskader (Kfr Medisinsk støttebehandling kap. 10) (Wright, Green, & Daw, 2009).
Tilbakefall av nefroblastom
I den nye Umbrella 2016-protokollen har RTSG sammen med COG utarbeidet retningslinjer for behandling av tilbakefall av nefroblastom. Tilbakefallene er sjeldne i en sykdom med så god prognose. De representerer likevel store utfordringer, og økt behandlingsintensitet medfører også risiko for langtidseffekter av behandlingen.
Her brukes som basis de samme medikamentene, vinkristin og actinomycin D (Spreafico et al., 2009) som i primærbehandling med tillegg av andre medikamenter som cyklofosfamid, etoposid og carboplatin. I noen tilfeller anbefales høydosebehandling med stamcellestøtte. I tillegg må mange ha strålebehandling.
Behandling av nefroblastomatosepasienter
Disse pasientene er i økt risiko for å utvikle nefroblastom og skal ha spesielt oppfølgingsprogram og behandling. Det gis ofte langvarig kjemoterapi (minst ett års varighet) inntil forandringene forsvinner eller inntil det blir mulig å fjerne dem med kirurgi. Etter en slik operasjon må det også gis langvarig kjemoterapi.
Andre nyresvulster
I tilegg til en rekke benigne svulster kan det oppstå andre maligne svulster i nyrene; Klarcellet nyresarkom (CCSK), «malign rhabdoid tumour of the kidney» (MRTK), nyrecarcinom samt metastaser fra annen kreftsykdom.
Øvrige retningslinjer som finnes i protokollen
- Behandling for pasienter med primær kirurgi.
- Behandling av bilateralt nefroblasom (gjelder ca 5 % av pasientene).
- Opplegg for behandling av nefroblastomatose.
- Behandling av clear cell sarcoma of the kidney, rhabdoid tumour of the kidney og residiverende sykdom.
- Retningslinjer for oppfølging.
- Behandling av nefroblastom hos voksne.
Kirurgisk behandling av nefroblastom
Det er enighet i det norske barneonkologiske miljøet om nasjonal deltakelse i Umbrella-protokoll SIOP-RTSG 2016, og studien er godkjent av etisk komite. Studien er fortsatt ikke åpnet for inklusjon av norske pasienter, men som «best available treatment» behandler vi i henhold til denne selv om pasientene ikke kan inkuderes i studien. Behandlingen er altså risiko-adaptert og det er viktig å følge protokollen i dens retningslinjer både for kirurgien, stadiesettingen og rapporteringen (sistnevnte kun når pasienter inkluderes). Behandlingsteamet må kjenne behandlingsprotokollen og ha meldeskjemaene for kirurgi for hånden før og/eller under operasjonen.
Tumor i én nyre
Ved operasjon for Wilms’ tumor prioriteres god tilgang fremfor kosmetisk, begrenset snitt. Det anbefales bred, transversal incisjon ovenfor umbilicus og tilgang via laparatomi. Minimal invasiv kirurgi (kikkhull/laparoskopi) er så langt ikke aktuelt hos barn. Bukhulen inspiseres og palperes nøye med tanke på spredning. Moderne billedfremstilling preoperativt gjør det unødvendig å åpne peritoneum/nyrefascien over motsatt sides nyre hvis det ikke er påvist suspekte forandringer i den.
Et generelt prinsipp ved tumorkirurgi er å benytte såkalt «non touch / minimal touch»-teknikk før drenerende kar fra tumor er avstengt. Om mulig bør arterien avklemmes først. Binyren fjernes sammen med nyren hvis ikke tumor er i god avstand fra den. Lymfeknute-sampling er viktig, kfr. behandlingsprotokollen, men radikalt lymfeknutetoilette er (for tiden) ikke indisert. Hvis det foreligger gjennomvekst av kapsel, må en sørge for god margin ut i normalt vev. Heroiske og mutilerende inngrep anbefales imidlertid ikke siden de fleste nyresvulster er både kjemo- og strålesensitive. Preoperativ cytostaticabehandling reduserer sjansene for kapselruptur ved uttak av tumor, men kan vanskeliggjøre bedømmelsen av tumorgrense p.g.a. nekrose/inflammasjon. NB! Sjekk tumors utbredelse på bilder tatt før behandling!
Tumortrombe i vena renalis eller vena cava skal fjernes via venotomi; om tromben når til atriet kan cardiopulmonal bypass være nødvendig.
Nefronsparende tumorreseksjon i stedet for radikal nefrektomi anbefales foreløpig ikke ved ensidig tumor.
Bilateral nyretumor
Ved bilateral tumor bør «nefronsparende» operasjon tilstrebes. Helt avhengig av tumors utbredelse står valget mellom lokal tumorexcisjon bilateralt eller nefrektomi på den ene siden og begrenset reseksjon på den minst affiserte siden. Enukleasjon av små tumores kan være aktuelt, spesielt hvis det er multiple slike i en nyre.
Fjernmetastaser
Lokale levermetastaser skal excideres i forbindelse med primæroperasjonen. Ved utbredte metastaser i leveren skal det tas biopsi. Lungemetastaser skal excideres, men inngrepet kan med fordel utsettes et par uker til resultatene av primæroperasjonen foreligger. Følg for øvrig gjeldende behandlingsprotokoll.
Nevroblastom
Nevroblastom er en ondartet svulst med utgangspunkt i embryonale perifere nerveceller som vanligvis diagnostiseres hos små barn, men sporadiske tilfeller kan også forekomme hos ungdom og unge voksne. I Norge diagnostiseres 6–10 nye tilfeller årlig, og rundt halvparten opptrer som metastatisk sykdom. De fleste nevroblastomer har utgangspunkt i binyrene, men svulsten kan oppstå alle steder der det er sympatisk nervevev (dvs grensestreng og ganglier). Prognosen ved nevroblastom varierer fra nær 100 % overlevelse ved lokal sykdom og gode prognostiske faktorer til rundt 50 % overlevelse hos barn over 1 år med metastatisk sykdom. Dårligst prognose finner vi hos barn > 18 mnd på diagnostidspunktet. Av den grunn er det stor variasjon i behandlingsintensitet hos barn med nevroblastom, fra kun observasjon i de enkleste tilfellene til multimodal intensiv terapi med cellegift, kirurgi, høydosebehandling med stamcellestøtte, stråling og immunterapi hos barn over 1 år med metastatisk sykdom. I Norge behandles nærmest alle barn med nevroblastom etter SIOPEN-protokoller. I SIOPEN samarbeider en rekke land for å oppnå de beste vitenskapsbaserte behandlings- og forskningsprotokoller. Norge er et aktivt medlem av SIOPEN, sammen med 24 andre medlemsland (https://www.siopen.net/).
Diagnose
Symptomer ved nevroblastom er svært varierte og inkluderer skjelettsmerter, fraktur, økt bukomfang, høyt blodtrykk, svette, diaré, nevrologiske utfall, periorbital blødning, Horner syndrom og påvirkning av perifere hematologiske verdier. Diagnosen stilles ved biopsi/cytologi av tumor eller benmargsmetastaser. Forhøyede urinkatekolamin-metabolitter (HVA og VMA), NSE og positive funn ved den nukleærmedisinske undersøkelsen mIBG styrker mistanken om nevroblastom før den cytologiske/histologiske diagnosen foreligger. PET-CT kan gi supplerende informasjon, men anses ikke som nødvendig ved positiv mIBG scan. Det er viktig å sikre nok tumorvev for biologiske analyser (f.eks MYCN-amplifikasjon, kromosomale delesjoner og gains i tumorvev) som kan predikere prognose og nødvendig behandlingsintensitet.
Radiologisk utredning
Siden klassifiseringen baserer seg på den radiologiske utredning med MR og ultralyd (Monclair et al., 2009), er det svært viktig at de preoperative undersøkelsene utføres ved det behandlende sykehus, etter faste protokoller, slik at sammenlikningen av undersøkelsene blir enklere.
Klassifisering
Tidligere stadieinndeling 1–4(S) er med nye protokoller blitt erstattet av den internasjonale INRG-klassifiseringen som forholder seg til risikofaktorer for kirurgi, metastaser og alder (Cohn et al., 2009; Monclair et al., 2009; Pinto et al., 2015);
| L1: | operabel tumor bedømt ut fra radiologiske risikofaktorer |
| L2 | inoperabel tumor bedømt ut fra radiologiske risikofaktorer |
| M: | metastaser til stede |
| MS: | barn under 1 år (18 mnd) med metastaser kun lokalisert til hud, lever og/eller benmarg |
Ung alder og gunstig histologi er forbundet med god prognose, mens biologiske faktorer som segmentale kromosomale aberrasjoner i tumorceller har størst betydning for utfallet av kreftsykdommen. I denne sammenheng er N-Myc amplifikasjon, 11-q delesjon, 1-p delesjon og 17-q gain de viktigste kromosomale avvik. N-Myc amplifikasjon er vanligst hos de minste barna, mens 11-q delesjon har størst betydning for utfallet ved nevroblastom hos eldre barn. Denne erkjennelsen har medført at biologiske faktorer tas hensyn til ved klassifisering og planlegging av behandlingen ved nevroblastomsykdom hos barn.
Behandling
Intensiteten i behandlingen av nevroblastom varierer avhengig av prognostiske faktorer (Pinto et al., 2015). Ytterpunktene demonstreres ved at man ved stadium MS uten livstruende symptomer eller MYCN-amplifikasjon kun observerer, mens man ved stadium M tar i bruk all tilgjengelig målrettet medisinsk behandling (kjemoterapi, kirurgi, høydosebehandling med stamcellestøtte, stråling, vitamin A og immunterapi med spesifikke GD-2 antistoffer). Kjemoterapien består av cisplatin, karboplatin, cyklophosfamid og vinkristin i induksjonsfasen, evt supplert med topotecan, doksorubicin, temozolomid eller irinotecan dersom induksjonsbehandlingen ikke har tilstrekkelig effekt. Nevroblastom har blitt oppfattet som en kreftform der kirurgien er avgjørende. Det er fortsatt tilfelle, men oppdagelsen av biologiske risikofaktorer har medført at enkelte «gunstige» svulster kan observeres uten kirurgi i første omgang. Følgende SIOPEN-protokoller brukes ved de ulike stadier:
| L1: | ingen åpen studie protokoll, kun kirurgisk behandling |
| L2: | protokoll SIOPEN LINES |
| M: | HR-NBL-1 SIOPEN |
| MS: | protokoll SIOPEN LINES / HR-NBL-1 SIOPEN ved MYCN-amplifikasjon |
Internasjonalt er bruken av immunterapi med GD-2 antistoff økende med lovende resultater (Yu et al., 2010). Dinutuximab Beta ble godkjent i Beslutningsforum juni 2019 til behandling av høyrisiko nevroblastom og relapserende/refraktær nevroblastom.
Ved høyrisk nevroblastom kombineres all tilgjengelig effektiv terapi i første behandlingsrunde. Av den grunn er det per i dag lite dokumentert behandling tilgjengelig i residivsituasjoner. Forskningen er intens på dette fagfeltet, og man har håp om at nye behandlingsformer blir dokumentert effektive i nær framtid. Høydose MIBG-behandling med eller uten topotekan-sensitivisering (Gaze et al., 2005) har vist seg effektiv i enkelte situasjoner og kan nå tilbys i Norge i en residiv- eller refraktær situasjon. For høyrisiko nevroblastomer i en residiv/refraktær situasjon der det foreligger informasjon om en ALK abberasjon i tumor, kan det være indisert med behandlingsforsøk med ALK-hemmer (fortrinnsvis en første generasjons ALK hemmer, men noen spesifikke ALK abberasjoner har kjent resistens mot første generasjons ALK hemmere). Denne behandlingen bør fortrinnsvis kombineres med annen tumorrettet behandling. ALK hemmer inngår per 2020 i førstelinjebehandling av høyrisk nevroblastom med ALK abberasjon i USA (Childrens Oncology Group protokoll ANBL 1531). I Europa har man i første omgang ikke inkludert dette i primærbehandlingen, men det brukes i utstrakt grad i en residivsituasjon.
Kirurgi ved nevroblastom
Kirurgisk behandling av nevroblastom vil som regel være radikal fjernelse av tumor. Dette bør skje uten mutilering, skade på vitale organer eller alvorlig annen morbiditet. For svulster med gunstige biologiske markører kan det være akseptabelt å la en liten resttumor bli værende igjen.
Kirurgi er lokalbehandling. Kirurgiens plass i behandlingen må derfor vurderes ut fra om det kan oppnås radikal fjernelse eller ikke. Det er viktig med god kartlegging av tumors utbredelse, både ved diagnosetidspunkt og preoperativt.
Selv om mange nevroblastomsvulster er svært følsomme for kjemoterapi, vil man med unntak av enkelte spedbarn, ikke oppnå at primærtumor forsvinner helt.
Preoperativ kjemoterapi vil ofte være aktuelt, både for å sanere metastaser og for å øke muligheten for radikal fjernelse av tumor. Ved tilstedeværelse av risikofaktorer bør det gis preoperativ kjemoterapi. Ved metastatisk sykdom vil det også gis preoperativ kjemoterapi selv om primærtumor er uten risikofaktorer, med mindre radikal fjernelse av primærtumor anses å være det beste alternativet for vevsdiagnostikk.
Preoperativ kjemoterapi gjør svulstene mindre, mer fibrøse, mindre vaskulariserte og generelt lettere å håndtere. Dette reduserer risiko for spredning peroperativt, og det er akseptabelt å dele tumor under operasjonen og ta den ut bitvis. Det er ofte nødvendig å dele tumor peroperativt fordi tumor ofte omslutter kar som må bevares. Svulstene kan være kraftig adherente til karveggen, men infiltrasjon i selve karveggen er svært sjelden. Etter kjemoterapi er restsvulsten ofte dårlig definert, og det er umulig peroperativt å skille mellom aktivt nevroblastomvev og modent ganglionevrom, fibrose eller arrvev. Derfor vil man ved operasjonen ha som målsetning å fjerne vev i primærtumors utbredelsesområde, men unngå å skade vitale strukturer. Det vanligste er å operere med åpen kirurgi, men for enkelte lokaliserte svulster kan det være aktuelt med laparoskopi eller torakoskopi.
Komplikasjoner relatert til nevroblastomoperasjoner avhenger av utbredelse og lokalisasjon. Ved inngrep på hals og øvre thorax kan det oppstå nerveskader (eks Horners syndrom). I nedre thorax er det en viss risiko for skade på medullas blodforsyning. Retroperitoneale operasjoner kan gi postoperativ diare og ascites. Ved ekstirpasjon av sentrale abdominale nevroblastomer er det risiko for nyreischemi. Nevroblastomkirurgi i bekkenet har risiko for nerveskade, både til blære, tarm og underekstremiteter.
Toksisitet/seneffekter etter nevroblastombehandling
Metastatisk høyrisk nevroblastom krever intensiv behandling, og dermed er risikoen høy for akutt toksisitet og alvorlige seneffekter både forårsaket av grunnsykdommen og behandlingen. Nevroblastomets lokalisering øker ytterligere risikoen for varige seneffekter, siden svulsten ofte infiltrerer nyre eller vokser inn i spinalkanalen. Likevel er det behandlingsinduserte seneffekter av hørsel og nyrefunksjon som er de vanligste følgetilstandene hos langtidsoverlevere etter intensivt behandlet nevroblastom.
Oppfølging/kontroller
Pasientene følges i minst 10 år etter endt behandling. Tumorovervåking baserer seg på ultralyd av tumorlokalisasjon (evt. MR dersom området ikke er tilgjengelig med ultralyd) og urinkatekolaminer. Radiologisk oppfølging av primærtumor kan avsluttes fem år etter endt behandling. Hørsel og nyrefunksjon er av de viktigste seneffektene å monitorere minst 1, 5 og 10 år etter gjennomført behandling.
Leversvulster
2/3 av leversvulstene i barnealder er ondartede. Dette er sjeldne tilstander og utgjør ca 1–2 % av barnekrefttilfellene. Totalt ser vi 0–3 nye tilfeller i Norge pr år.
90 % av de ondartede svulstene i lever er enten hepatoblastom (HB) eller hepatocellulært carcinom (HCC). HB forekommer langt hyppigere enn HCC (Ratio 1,8: 1)
Fordi svulstene er så sjeldne, er det på verdensbasis dannet samarbeidsgrupper som sammen utvikler programmer for diagnostisering og behandling samt forskning på disse svulstene.
I Norge er vi tilsluttet SIOPs levertumorgruppe SIOPEL (https://siope.eu/news/siopel-patientsfamilies-forum-liver-tumours-children/) som har ledet multinasjonale, kliniske studier på leversvulster hos barn siden 1990. Mer enn 30 land deltar i dette SIOPEL–samarbeidet.
Andre store grupper for behandling av leversvulster hos barn er:
- COG (Children’s Oncology Group), USA (www.childrensoncologygroup.org and www.curesearch.org)
- Levertumorgruppen i GPOH (German Society of Pediatric Hematology and Oncology), Tyskland/Østerrike.
- JCCG – The Japanese children’s Cancer group, Japan
Felles for HB og HCC er at de kan produsere og skille ut alfa-fetoprotein (AFP), et glykoprotein som syntetiseres i plommesekk og lever fra tidlig i fosterlivet. Produksjonen stopper normalt ved fødsel, og konsentrasjonen synker fra gjennomsnittlig 50 000 ng/mL ved fødsel til < 20 ng/mL ved 6–8 måneders alder. Variasjonsbredden i AFP-nivå er stor, og det anbefales at man slår opp i tabell i aktuell protokoll. Helt nede på voksne verdier er man først ved 1–2 års alder.
AFP er forhøyet hos over 90 % ved HB og hos vel 50 % ved HCC.
Hepatoblastom
HB er en ondartet svulst med utgangspunkt i embryonale leverceller. Median alder ved diagnose er 1 år. 80 % sees i de 3 første leveår. HB er også beskrevet opp i ungdoms- og voksenalder. HB opptrer oftest som en solitær leversvulst, men kan være multifokal og da affisere store deler av leveren.
Genetikk. HB opptrer oftest sporadisk, og der vet man foreløpig lite om genetiske og molekylærbiologiske faktorers betydning for utvikling, risikoinndelig, behandling og prognose. HB ses også i assosiasjon med visse syndromer hvor noe mer av genetikken er kjent.
Tilstander som disponerer for hepatoblastom
- Isolert hemihypertrofi
- Beckwith-Wiedemann syndrom
- Familiær adenomatøs polypose
Pasienter med disse syndromene/utviklingsforstyrrelsene skal kartlegges og tilbys jevnlig kontroll. Man skal også se nøye etter tegn på disse tilstandene ved diagnostisert HB.
Økt risiko for HB ses også som en assosiasjon til prematuritet med svært lav fødselsvekt.
Diagnose
HB presenterer seg oftest som en asymptomatisk, stor oppfylning i magen hos et ellers friskt barn. Noen har også allmennsymptomer i form av feber, vekttap, anorexi og anemi. Hos noen sees det også koagulasjonsforstyrrelser med blødning ved diagnose.
Laboratorieutredning viser ofte trombocytose og oftest forhøyet AFP-verdi. I sjeldne tilfeller kan også en annen tumormarkør, beta-chorionic gonadotropine (β-HCG) være forhøyet med økt risiko for tidlig pubertetsutvikling.
Utredning
- Grundig anamnese og generell klinisk undersøkelse
- Blodprøver med tumormarkørpanel (AFP, b-HCG, LD, koagulasjonsprøver, evt andre)
- Ultralyd abdomen
- MR eller CT av abdomen
- Rtg thorax og CT thorax
- Evt MR eller CT caput
- Urin til katekolaminbestemmelse (differensialdiagnostikk)
Diagnostisk biopsi
Biopsi gjøres nå som regel som ultralydveiledet tru-cut biopsi som utføres av radiolog i generell anestesi, og med patolog til stede for å sikre adekvat vevsmengde. Det kan også være nødvendig med kirurgisk biopsi hos noen, avhengig av tumors beliggenhet i leveren.
Biopsi er sterkt anbefalt for alle pasienter med mistenkt malign levertumor med mindre det foreligger blødningsforstyrrelser eller kritisk organpåvirkning.
Histologi
Hepatoblastomene deles inn i epiteliale undergrupper: Fetal, embryonal, makrotrabekulær og småcellet udifferensiert (tidligere kalt anaplastisk) og i blandet epitelial og mesenkymal morfologi med eller uten teratoide trekk.
Klassifisering og stadieinndeling
Det har tidligere blitt benyttet flere ulike systemer for klassifisering og stadieinndeling internasjonalt. I USA og flere andre land har det vært vanlig å gjøre primær kirurgi der dette er mulig, og deres inndeling og stadiesetting har vært basert på dette. Dette er forskjellig fra den europeiske tilnærmingen til behandling, noe som har gjort det vanskelig å sammenfatte og sammenligne forskning på dette feltet.
Den pågående studien på leversvulster som Norge også deltar i er imidlertid et samarbeidsprosjekt mellom den europeiske gruppen (SIOPEL), den amerikanske gruppen (COG) og den japanske gruppen (JCCG) kalt PHITT (paediatric hepatic international tumour trial), og man vil i denne studien undersøke kvaliteten på et felles klassifiseringssystem og behandling. Hovedmedikamentet som har vist seg svært effektivt på leverkreft er cisplatin. Dette gis ofte i kombinasjon med doxorubicin og noen ganger også alternerende med carboplatin. Klassifiseringen følger et skjema utviklet av SIOPEL hvor svulstene blir inndelt etter «PRE-Treatment EXTent of disease» (forkortet PRETEXT) Systemet baserer seg på radiologisk påvist affeksjon av leveren ved diagnose i forhold til leverens anatomi med inndeling i 4 seksjoner (8 leversegmenter) samt innvekst i kar og evt tilstedeværelse av metastaser(For beskrivelse av leveranatomi vises det til SIOPEL publikasjoner og protokoller) (Aronson et al., 2005).
Svulstene deles i:
PRETEXT I: kun 1 seksjon involvert
PRETEXT II: 2 «naboseksjoner» involvert
PRETEXT III: 2 seksjoner som ikke ligger inntil hverandre eller 3 «naboseksjoner» involvert
PRETEXT IV: alle 4 seksjoner involvert
Videre inndeles pasientene i very low, low, intermediate og high risk ut fra følgende faktorer:
Tumoregenskaper (ruptur, fokalitet, innvekst i blodkar, vekst utenfor lever, ruptur/blødning)
Metastaser
Barnets alder
AFP-verdi
Tumors resektabilitet
Maligne leversvulster metastaserer først og fremst til lunger og lymfeknuter, men det er også sett spredning til skjelett og CNS. Leversvulstene metastaserer ikke til benmarg.
Behandling
HB er fortsatt en kirurgisk sykdom. Noen få pasienter er primært operable, men strategien for de fleste risikogruppene er preoperativ kjemoterapi med utsatt kirurgi.
Mange pasienter kan få tumor fjernet med leverreseksjon etter preoperativ kjemoterapi. I tillegg kan enkelte, tidligere inoperable pasienter tilbys levertransplantasjon. Kjemoterapien totalt er med på å hindre tilbakefall for de aller fleste. Til sammen har dette gjennom 20 år gitt en betydelig bedret prognose (Perilongo et al., 2009; Perilongo et al., 2004; Pritchard et al., 2000). Se også kapittelet om Kirurgi.
I Norge brukes PHITT-protokollen som klassifiserings- og behandlingsretningslinje, enten barnet deltar i studien eller ikke.
Overlevelse (overall survival) varierer med risikogruppe, og er for standard risiko HB 95 % og for høyrisikogruppen ca 80 % (Zsiros et al., 2013).
Hepatocellulært carcinom
HCC sees oftest hos litt eldre barn (median alder ved diagnose 12 år) og er en langt alvorligere tilstand. HCC forekommer hyppigere i områder hvor hepatitt B virus (HBV) er endemisk, og da oftest hos barn over 5 år. HCC er også assosiert med hepatitt C virus (HCV)
Tilstander som disponerer for HCC
Underliggende leversykdommer som bl.a. tyrosinemi, biliær atresi, idiopatisk neonatal hepatitt, a1‑antitrypsinmangel, nevrofibromatose, ataxia teangiectasia er assosiert med HCC.
Diagnose
Klinisk bilde som ved HB, men positiv AFP kun hos ca 50 %.
Utredning
Som ved HB. I tillegg utvidede tester med fokus på underliggende leversykdom.
Diagnostisk biopsi
Biopsi (ultralydveiledet) bør gjøres hos alle med mindre tumor kvalifiserer for primær kirurgi.
Histologi
Hepatocellulære carcinomer deles histologisk i pediatrisk, adult og fibrolammelær HCC.
Klassifisering og stadieinndeling
Inndeling i PRETEXT-grupper som ved HB – se over.
Maligne leversvulster metastaserer først og fremst til lunger og lymfeknuter, men det er også sett spredning til skjelett og CNS. Leversvulstene metastaserer ikke til benmarg
Behandling
Behandling av HCC er en utfordring, og suksess i behandlingen er totalt avhengig av mulighetene for kirurgi. Dersom tumor er operabel, skal pasienten ha primær kirurgi da HCC er en relativt kjemoresistent tumor. Under halvparten er operable ved diagnose. Pasienter som ikke er operable ved diagnose eller som har mikroskopisk restsykdom etter diagnose vil randomiseres til å motta en av to ulike kjemoterapiregimer. Målet med dette er å finne en behandling som gir reduksjon i tumor slik at flere blir operable, enten ved reseksjon eller ved transplantasjon, og at flere pasienter blir kurert. Tilbakefall av sykdom ved HCC er også en stor utfordring til dags dato.
Generelle kriterier for levertransplantasjon har til nå vært strenge. Hos voksne pasienter aksepteres ikke svulster med diameter > 5 cm eller multifokalitet med mer enn 3 svulster, spesielt ikke hvis en av dem er > 3 cm eller kombinert diameter er > 8 cm (Milan criteria), men man gjør oftest en mer individualisert vurdering for barn. Dog er begrenset sykdom fortsatt et kriterium for suksess også ved transplantasjon. Metastatisk sykdom vil være kontraindikasjon for transplantasjon.
5 års overall survival for HCC i tidligere SIOPEL-protokoller har vært under 30 %. Man har til nå brukt de samme medikamenter ved HCC som ved hepatoblastom, men uten like stor suksess. Man er nå inne i en fase med mer eksperimentell medikamentell behandling i håp om et gjennombrudd for disse pasientene. I PHITT-protokollen sammenlignes behandling med Gemcitabin og Oxaliplatin med konvensjonell HB-behandling. I tillegg er det lagt til Sorafenib i behandlingen for alle pasientene. Dette er et medikament som har vist effektivitet hos voksne pasienter med HCC. I senere studier kan aktuelle medikamenter være bevacizumab, everolimus, sirolimus osv. Lokal intervensjons-behandling direkte mot tumor med radioembolisering eller medikamenter kan også bli aktuelt.
Ved HCC skjer behandlingen etter individuell vurdering og rådføring med internasjonalt fagmiljø.
Overlevelse (overall survival) for HCC er rundt 50 % etter kirurgisk reseksjon/transplantasjon.
Oppfølging
Som for de fleste andre solide svulster følges også barn etter leversvulstbehandling i minst 10 år etter avsluttet behandling og gjerne gjennom puberteten. Barna følges dels for å avdekke residiv, men like viktig er oppfølging med tanke på senskader etter behandlingen.
De to første årene kommer barna til radiologisk undersøkelse hver tredje-fjerde måned. De fleste greier seg med ultralyd abdomen og røntgen thorax, mens noen må ta MR/CT. Man forsøker å unngå CT i kontrolloppfølging grunnet stråledosen. Etter 2 år blir det gradvis lengre intervall mellom kontrolltimene, og vanligvis avsluttes radiologisk oppfølging etter 5 år.
Barn som har fått store kumulative doser kjemoterapi (Cisplatin, Doxorubicin) som kan skade hjerte, nyre eller hørsel får relevante undersøkelser 1, 5 og 10 år etter behandlingsavslutting (kardiotox, hørselstest, GFR).
Kirurgisk behandling av maligne leversvulster
Behandlingen av leverkreft foregår i tett samarbeid mellom barneonkologer, barnekirurger og transplantasjonskirurger. Komplett fjerning av tumor med frie reseksjonsrender er kritisk for langtidsoverlevelse for både hepatoblastom og hepatocellulært carcinom. Leveren har stor regenerasjonsevne, og reseksjoner opp mot 80 % kan tolereres. Hepatoblastomer vil oftest være kjemosensitive, og preoperativ kjemoterapi kan gjøre kirurgien tryggere. Noen svulster kan ved diagnosetidspunktet gi inntrykk av å involvere aller fire segmenter i henhold til Pretext-inndeling. Etter kjemoterapi vil man oftest se at svulsten kan fjernes. Ved multifokale svulster og Pretext IV-svulster vil levertransplantasjon være aktuelt. For enkelte sentralt beliggende svulster som ikke kan fjernes, kan også levertransplantasjon være et alternativ. Komplikasjoner relatert til leverreseksjoner er blødning, gallegangsskader, karskade, luftemboli og leversvikt.
Germinalcellesvulster utenfor CNS
Germinalcellesvulstene (GCT) er en svært heterogen gruppe svulster som varierer betydelig i klinisk presentasjon, lokalisasjon, histologi og biologi. Hos ungdom og unge voksne er de oftest lokalisert i gonadene. I barnealder derimot oppstår de oftest ekstragonadalt i midtlinjen, f eks sacrococcygealt eller i fremre mediastinum. GCT diagnostiseres i alle aldersgrupper fra fosterlivet og opp gjennom oppveksten og står for 3–4 % av krefttilfellene hos barn < 15 år. Modne teratomer kan være underrapportert i slike statistikker (Gobel et al., 1998; Gobel et al., 2000).
Histologi
Svært varierende mikroskopiske bilde. Det ses benigne tumorer, såkalte modne teratomer som kan bestå av mange ulike vevstyper. Det ses også svulster med større eller mindre malignt potensiale, såkalte maligne germinalcellesvulster (MGCT).
Revised Histological Classification of Germ Cell Tumours
- Germinoma
(1) Intratubular
(2) Invasive - Teratoma
a. Mature
b. Immature
c. Malignant teratoma (Teratoma with non germ cell malignant component) - Embryonal carcinoma
- Yolk sac tumour
- Choriocarcinoma
- Gonadoblastoma
- Mixed malignant germ cell tumour
Genetikk
Tilstander som disponerer: Klinefelter syndrom disponerer for mediastinal MGCT.
Ovariale GCT’er viser en signifikant assosiasjon med konstitusjonelle forandringer i kjønnskromosomer som f.eks ved Turner syndrom.
Diagnose
GCT presenterer seg oftest som en asymptomatisk, stor, plasskrevende oppfylning som kan presse på andre organer og gjennom det gi symptomer, for eksempel obstruksjon i luftveiene, vena cava-syndrom, obstipasjon ved bekkensvulster osv. GCT’er skiller ofte ut tumormarkører (AFP og/eller β‑HCG) avhengig av histologisk differensiering. Mønsteret av disse er med på å gi en klinisk diagnose. Husk at AFP normalt er svært høyt i nyfødtperioden og først er nede i «voksne verdier» ved 1–2 års alder. (Mer om AFP, se kapitlet om leversvulster.)
Utredning
- Grundig anamnese og generell klinisk undersøkelse.
- Blodprøver med tumormarkørpanel (AFP, β-HCG og LD. Evt PLAP (placenta-like alkanin fosfatase) som kan være positiv i serum ved germinomer)
- Evt kromosomanalyse (Klinefelter/Turner o.a.)
- Ultralyd abdomen
- MR (CT) av av aktuelle område hvor tumor er lokaliser og av abdomen
- Rtg thorax og CT thorax
- Urin til katekolaminbestemmelse (differensialdiagnostikk)
Klassifisering og stadieinndeling for MGCT varierer mellom ulike studiegrupper og deres protokoller. Protokollen vi følger, UKCCSG GC III, baserer seg på en vanlig TNM-klassifisering, og behandlingsstrategien deler svulstene i lav-, intermediær- og høyrisiko sykdom avhengig av lokalisasjon, utbredelse og nivåer av tumormarkørsekresjon – for detaljer, se protokoll.
De ulike nasjonale og flernasjonale gruppene har ennå ikke klart å samle seg på samme måte som for en del andre solide svulster. Det er derfor flere grupper med litt ulike studier i Europa samt større studier i regi av COG i USA. Det er imidlertid dannet en International Germ Cell Tumour Group som håper å komme i gang med felles studier hvor også randomiserte spørsmål kan bli besvart.
Vi følger nå den engelske behandlingsprotokollen for GCT – UKCCSG GC III (2005) og Children’s Cancer & Leukaemia Group, Germ Cell Group UK- ‘INTERIM GUIDELINES FOR THE TREATMENT OF EXTRACRANIAL GERM CELL TUMOURS IN CHILDREN AND ADOLESCENTS’ fra 2011. Det gjør også Sverige og Danmark, og vi samarbeider i NOPHOs arbeidsgruppe for GCT. I denne protokollen brukes carboplatin i motsetning til cisplatin som de fleste andre studier benytter. Carboplatin gir mindre skade på hørsel og nyrer enn cisplatin gjør, og carboplatin har vist seg i barnealder å være like effektivt som Cisplatin i de to foregående engelske studiene. Carboplatin gis da i kombinasjon med etoposid og bleomycin (J. R. Mann et al., 2000).
Behandling
Modne teratomer fjernes kirurgisk, men skal følges videre med kontroller (klinisk + ultralyd + markører) da de kan residivere både som modent teratom og som MGCT hvis kirurgien har vært inkomplett. Dette gjelder særlig sacrococcygeale svulster hos spedbarn hvor residivraten er på 10–14 %, og hvor residivene ofte er plommesekksvulster.
Immature teratomer. Kirurgi er hovedbehandlingen og er oftest tilstrekkelig alene. Betydningen av økt AFP ved immature teratomer er kontroversiell, og for tiden anbefales at immature teratomer med AFP> 1000 kU/L ved diagnose behandles som MGCT med kjemoterapi i tillegg til kirurgi. De med AFP < 1000 kU/L skal følges tett med ultralyd hver 3. måned og tumormarkørbestemmelse hver 4. til 6. uke. Ved gjenvekst av tumor anbefales ny kirurgi og deretter kjemoterapi.
Alle GCT’er skal ha målt AFP og HCG og gjennomført full utredning. Hvis metastaser ikke er påvist, skal kirurgi vurderes. Inoperable svulster skal, hvis mulig, biopseres. Mutilerende kirurgi skal unngås. Utsatt kirurgi kan vurderes, men skal fortsatt unngås hvis det medfører skader av betydning. GC II-studien har vist at bare 2 av 45 barn hadde viabelt tumorvev ved utsatt kirurgi etter gjennomført kjemoterapi. De øvrige hadde nekrose/fibrose eller modent/umodent teratom. Slike pasienter bør heller følges tett for å se om utviklingen nødvendiggjør ytterligere behandling.
Lavrisiko MGCT defineres som alle stadium 1 svulster (kfr protokoll) Disse får ingen postoperativ kjemoterapi, men følges tett med ukentlige kontroller til normalisering av tumormarkører og deretter kontroll hver måned.
Intermediær risiko MGCT er alle seminomer/germinomer uansett AFP-verdi, de fleste stadium 2 og 3 svulster med AFP < 10 000 kU/L unntatt thoracale svulster hvor st. 2, 3 og 4 alle er høyrisiko, mens testis st 4 < 5 år er intermediær.
Disse får 4 kurer med kjemoterapi.
Høyrisiko MGCT er alle med AFP > 10 000 kU/L unntatt stadium 1 svulster og untatt testissvulster hos gytter < 5 år. Alle thoracale svulster st 2,3 og 4 inngår i gruppen uavhengig av AFP nivå.
Disse får 6 kurer med kjemoterapi.
Strålebehandling er ikke en integrert del av behandlingsopplegget for ekstrakraniell GCT i barnealder. Men strålebehandling kan være aktuell ved tilbakefall. Dysgerminom i ovariet og seminom i testis er svært strålefølsomme, og strålebehandling kan være aktuelt i utvalgte situasjoner. Men dette vil være etter individuell vurdering hos erfaren stråleonkolog.
Residivbehandling
Protokollen gir også anbefalinger for behandling ved residiv. Kjemoterapien er da vesentlig cisplatinbasert.
Prognose
Prognosen er generelt god. Gjennom de siste 20 år har prognosen for maligne GCT’er bedret seg betydelig. Mye av dette kan tilskrives bedret multimodal tilrettelegging av behandlingen samt ikke minst, introduksjonen av cisplatin i kjemoterapien. Der hvor man kan fjerne tumor radikalt, er overlevelsen nå > 80 %, men enkelte undergrupper som choriocarcinom kan ha dårligere prognose.
Oppfølging
De fleste barn følges i minst 10 år etter avsluttet behandling og gjerne gjennom puberteten. Barna følges dels for å avdekke residiv da disse ofte kan behandles med godt resultat. Like viktig er også oppfølging med tanke på senskader etter behandlingen.
Den første tiden følges de med forhøyede tumormarkører tett for å se at disse faller til normale verdier. Deretter månedlige tumormarkørkontroll i et år, så hver annen måned det andre året og hver 3. måned det tredje året.
De to første årene kommer barna også til radiologisk undersøkelse hver tredje måned. De fleste greier seg med ultralyd abdomen og røntgen thorax, mens noen må ta MR/CT. Man forsøker å unngå CT i kontrolloppfølging grunnet stråledosen. Etter 2 år blir det gradvis lengre intervall mellom kontrolltimene, og vanligvis avsluttes radiologisk oppfølging etter 5 år.
Barn som har fått store kumulative doser kjemoterapi som kan skade hjerte, lunger, nyre eller hørsel får relevante undersøkelser 1, 5 og 10 år etter behandlingsavslutting (kardiotox, lungefunksjonstest, hørselstest, GFR). Man er også oppmerksom på stråleinduserte skader og samarbeider med ortoped, pedagog og andre dersom tegn til stråleskader oppdages. Man er også oppmerksom på endokrinologiske bivirkninger og samarbeider tett med barneendokrinolog, eventuelt gynekolog.
Retinoblastom (RB)
Epidemiologi
Retinoblastom er en sjelden svulst som oppstår i ett eller begge øyne fra umodne retinaceller hos barn. Insidens av RB i Norge er omtrent 1/15 000 levende fødte barn (Gregersen et al., 2016). Det oppdages ca. 4 nye tilfeller per år og det er like mange gutter som jenter som får RB. Selv om RB kan inntreffe i alle aldre, oppstår det oftest før 2 års alderen, og ca. 95 % av tilfellene er diagnostisert før 5 års alder. Opp mot 98 % av alle barn med RB blir helbredet. Dette er et resultat av tidlig diagnostikk og gode behandlingsmuligheter.
Klassifikasjon
Nå klassiffiseres RB hovedsakelig i henhold til den Internasjonale klassifiseringen; ABCDE (Murphree, 2005). Tidligere benyttet man også Reese-Ellsworth (Yousef et al., 2015), og TNM (Rosengren, Monge, & Flage, 1989). Det kan være en fordel å benytte alle tre klassifikasjoner slik at det kan gjøres sammenlikninger med tidligere publiserte materialer.
Veksttyper
Endofytisk: Vokser fra netthinneoverflaten og inn mot eller inn i glasslegemet. Tumorens overflate er oppblåst og dårlig avgrenset med atypiske kar (figur 1).
Eksofytisk: Vokser utover og løfter opp netthinnen til evt. amotio (figur 2).
Diffust infiltrerbar: Dette er en uvanlig form som ofte diagnostiseres sent på grunn av differensialdiagnostiske vanskeligheter. De aller fleste pasientene er eldre enn ved de to andre formene. Tumor er vanligvis ensidig og oppstår spontant (Kim, Abramson, & Dunkel, 2007).
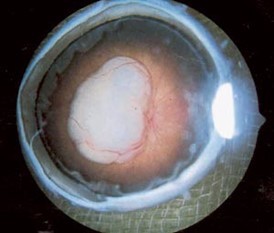
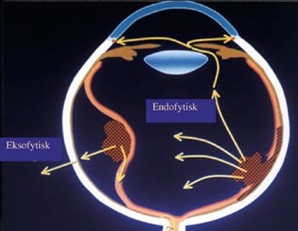
Patogenese, genetikk og etiologi
Retinoblastom skylles mutasjon av begge alleler av RB1 genet, som er lokalisert på kromosom 13 bånd q14 og er et stort gen som omfatter 200 000 basepar (Friend et al., 1986; Kohno, Kitajima, Sasaki, & Takahashi, 2016). RB1 er et tumor-suppressorgen. Pasienter med den arvelige formen har en medfødt germline mutasjon på et av allelene (first hit), som enten kan være nyoppstått eller nedarvet fra foreldrene (10 %). Penetransen ved RB1 mutasjoner er høy, men det finnes tilfeller med nedsatt penetrans (samlet penetrans er ca. 95 %). Tumor oppstår som en somatisk mutasjon i det andre allelet (second hit) (Cook et al., 2017). De fleste av de arvelige tilfellene er multifokale og bilaterale, og gjennomsnittsalderen ved diagnose er 15 mnd. Ved de ikke arvelige, sporadiske tilfellene er tumor oppstått som følge av to somatiske mutasjoner. Disse er oftest unilaterale og unifokale, og gjennomsnittsalderen ved diagnose er ca. 24 mnd.
Ved den arvelige varianten kan det også forekomme tumor i corpus pineale. Dette kalles trilateralt retinoblastom, og MR cerebrum us. er viktig for å utelukke dette.
Symptomer og tegn
Ved undersøkelse av barn, både ved nyfødtscreeningen og på helsestasjonene, bør den minste mistanke om leukokori eller skjeling medføre umiddelbar henvisning til øyelege (Dimaras et al., 2012; Sandven-Thrane, 2012). De vanligste debutsymptomene er:
- Leukokori (hvit refleks): 56–69 %
- Strabisme (skjeling): 20–35 %
- Nedsatt syn
- Glaukom
- Rødt øye
- Uveitter
- Unilateral mydriasis
Utredning
Øyeundersøkelse i våken tilstand: Sjekk av visus, synsfelt, skjeling, pupillerefleks og øyebevegelse.
Øyeundersøkelse i full narkose: Undersøkelse av øyebunnen i maksimal mydriasis og med oraimpresjon. Bruk av ultralyd for måling av tumorstørrelse, vurdere innhold av kalk og ev. gjennomvekst av øyeveggen. Klinisk foto av øyebunnen. Biopsi er ikke anbefalt p.g.a. risiko for sekundær spredning.
Blodprøver: Hb, leukocytter med differesialtelling, trombocytter, natrium, kalium, kreatinin, ASAT, ALAT, LD, CRP, leverfunksjonsprøver, serologiske undersøkelser for CVM, EBV og Varicella og prøver til genetikk.
Full somatisk undersøkelse.
MR orbita og cerebrum: Framstiller godt tumor fra vevet omkring. Ser etter mulig ekstraokulær vekst og corpus pinealesvulster. Tumorvevet fremstilles som moderat hyperintensivt på T1 og hypointensivt på T2 bilder. MR kan også skille mellom tumorvev, blødning og eksudat.
Barnet og familien henvises til genetisk samtale/veiledning.
Ev. CT orbita: Fremstiller kalk i tumor, god på påvisning av vekst utenfor øyet.
Ved ev. enukleasjon: Ta ferskt tumorvev til mutasjonsanalyse før formalinfiksering.
Behandling
Innledning
Både diagnostikk og behandling av retinoblastom er multimodal og omfattende og skal gis ved sykehus med spesialkompetanse i slik behandling.
Øyeavdelingen og Barneavdeling for kreft og blodsykdommer ved Oslo Universitetssykehus har landsfunksjon for retinoblastom. Alle retinoblastomene utredes og behandles primært her. Alle oftalmologiske kontroller under behandlingen gjøres her.
Små svulster kan behandles med lokalbehandling, som kryobehandling, laser behandling eller stråleplate, utført ved øyelege. Ved større svulster kan det være aktuelt med kjemoterapi, og i noen tilfeller må man fjerne øyet (enukleasjon). Ekstern stråling brukes sjelden i dag.
Om det skal gis konvenskonell, intravenøs kjemoterapi, kan dette gis på lokalsykehus med kompetanse på kjemoterapi. Mellom kurene kan barnet gjerne være hjemme, men må da ha tett kontakt med lokalsykehuset for blodprøvekontroller. Støttebehandling i form av transfusjoner og eventuell antibiotikabehandling, gis på lokalsykehuset etter vanlige retningslinjer.
Kjemoterapi
Ved behov for kjemoterapi, er primærbehandlingen nå ofte cytostatika (Melfalan) direkte inn i a. ophthalmica (IAM). Dette har vist goderesultater hos de fleste (Radros et al., 2018). I tillegg unngår man bivirkningene som ved systemisk behandling. Denne behandlingen brukes som volumreduserende tiltak før annen behandling (Shields et al., 2004; Shields, Meadows, Shields, Carvalho, & Smith, 2001). Nødvendigheten av tilleggsbehandling vurderes ved øyeundersøkelse i narkose i tilslutning til hver 2. kur. Om ikke IAM behandling er aktuelt eller er kontraindisert (for eksempel hos de aller minste barna), kan det gis systemisk kjemoterpi med Vincristin, Carboplatin og Etoposid med 3 ukers mellomrom. Det gis vanligvis 3–6 kurer.
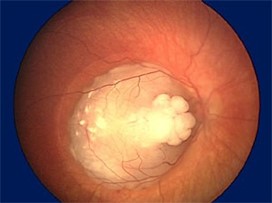
Kryobehandling
Vevet fryses til –70 °C. Benyttes ved mindre perifere svulster (mindre enn 6 mm i diameter og 3 mm høyde).
Laserbehandling
Det finnes ulike typer laser som benyttes for å ødelegge blodforsyningen til tumor. Benyttes ved små svulster (mindre enn 4–7 mm i diameter og 3 mm høyde) særlig de som ligger langt bak i øyet. Kan også gis i tilslutning til kjemoterapi primært, eller ved små residiv.
Strålebehandling
Retinoblastom er en strålefølsom tumor (Kim et al., 2007; Swanson et al., 2011)
Lokal strålebehandling med plate (Ru-106 (ruthenium) eller I-125 (jod)). Disse avgir betastråler respektive lavenergetiske gammastråler.
Indikasjoner:
Tumor < 15 mm i diameter og 3–7 mm tykk.
Tumores som er minst 2 mm fra n. opticus eller macula.
Tumores med lokal infiltratrasjon i glassvæsken.
Turmores som ikke har svart på annen behandling eller ved residiv.
Dosen er 35–40 Gy mot tumors apeks.
Ekstern strålebehandling
Indikasjon
Ved residiv hvor man vil bevare et seende siste øye, hvor annen behandling ikke har virket, eller ved spredning ut i glasslegemet.
Dosen er 35–40 Gy, 1,5–2 Gy 5 dager per uke i 5 uker.
Om det er aktuelt med strålebehandling, gis dette i form av protonbestråling som har kortere strålerekkevidde og derfor ikke skader tilstøtende vev like mye som fotoner.
Enukleasjon
Benyttes enten når svulsten ved diagnosen er så stor at det ikke er mulig å bevare visus, ved residiv man ikke får under kontroll, eller ved tegn til metastaser. Ved enukleasjon er det viktig at man får med seg så mye av n.opticus som mulig. Dersom det ved histologisk undersøkelse av det fjernede øye finnes tegn til gjennomvekst i n. opticus, eller ved tegn til infiltrasjon gjennom sklera, er det nødvendig med kjemoterapi eller strålebehandling (Nordiske retningslinjer for retinoblastom, 2016).
Prognose
Overlevelsen av RB er 98 %. Lokalt residiv er vanlig, men kommer svært sjelden etter at barnet har fylt 3 år.
Prognostiske faktorer mhp residiv:
Tumors størrelse og utbredelse.
Eventuelt forhøyet intraokulært trykk ved diagnosen.
Alder ved diagnose. Prognosen med henblikk på residiv er bedre ved diagnose mellom 2 og 7 år enn for pasienter som diagnostiseres tidligere eller senere.
Risiko for nye retinale tumores er (Abramson, Greenfield, & Ellsworth, 1992)
58 % hvis diagnosen stilles < 3 mnd alder
45 % hvis diagnosen stilles mellom 3 og 6 mnd alder
14 % hvis diagnosen stilles > 6 mnd alder
De som har arvelig retinoblastom, har større risiko for å utvikle sekundære maligniteter, dvs. sarkom i ben og bløtvev og kutane melanomer (Kleinerman et al., 2005; Kleinerman et al., 2012; Turaka, Shields, Meadows, & Leahey, 2012; Wong et al., 2014). 70 % av tilfellene oppstår i hodet (innen strålefeltet) ved ekstern strålebehandling Dette er avhengig av dose/respons og alder. Disse tumortypene andre steder i kroppen er ikke relatert til strålebehandling, men til mutasjonene i RB1-genet.
Utvikling av ev. komplikasjoner i øyet
Fra gjenværende tumor og arrvev
Vaskulære anomalier
Blødninger
Forandringer på retina
Glaukomutvikling
Ofte utvikling av pussdannelse ved enukleasjon.
Forandringer etter strålebehandling
Kosmetiske
Sekundær atrofi av orbita
Atrofi av fett og bindevev – øyet blir innsunket
Stråleretinopati og opticusnevropati
Utvikling av katarakt
Ofte store plager med tørre øyne, trenger kunstig tårevæske
Videre kontroller
Barn som er behandlet for RB bør gå til hyppige kontroller både hos pediater og oftalmolog med henblikk på:
Residiv
Ny cancer, primær eller sekundær (i strålefeltet)
De bør også følges opp på lokal BUP ved behov.
Øyelegekontroll:
Barn som har gjennomført behandling for retinoblastom bør kontrolleres:
Hver 2. mnd et år etter siste behandling. Deretter individuell nedtrapping. Avhengig av antatt arvelighet og gjenværende tumorforandringer i øyet/øynene skal noen ha kontroll hos øyelege hele livet.
Barn til foreldre med bilateralt retinoblastom bør kontrolleres:
Så raskt som mulig etter fødsel (innen maks 2–3uker)
Hver 2.–3. mnd til 2 års alder.
Hver 4. mnd til 4 års alder
Hver 6. mnd til 7–8 års alder
Søsken til barn med retinoblastom og barn til foreldre med unilateralt retinoblastom bør kontrolleres:
Hver 2.–3. mnd første året.
Hver 4. mnd til 4 års alder.
Hver 6. mnd til 7–8 års alder
Sjeldne svulster
Sist faglig oppdatert: 26.05.2020
I tillegg til de svulstene som her er omtalt, finnes det en rekke andre svulsttyper som forekommer svært sjelden. Dette gjelder dels svulsttyper som er mer eller mindre spesifikke for barnealder, dels mer «voksne» svulster. Felles for disse ekstremt sjeldne svulsttypene er at det ikke finnes spesifikke behandlingsprotokoller for barn, og man må som regel i hvert enkelt tilfelle skreddersy en behandling ut fra histologisk type, barnets alder og egne eller andres erfaringer med lignende svulsttyper.
Leukemier, lymfomer og histiocytoser
Sist faglig oppdatert: 26.05.2020
Leukemier
Det diagnostiseres hvert år ca. 35–40 nye tilfeller av leukemi hos barn under 15 år i Norge (Nasjonalt kvalitetsregister for barnekreft, 2015). De fleste, 80–85 %, er akutt lymfatisk leukemi (ALL), 10–15 % er akutt myelogen leukemi (AML), mens et fåtall utgjøres av andre, sjeldne leukemityper («voksen» type kronisk myelogen leukemi – KML med Philadelphia-kromosom, juvenil myelomonocyttleukemi – JMML). Heldigvis har prognosen for de vanligste leukemiformene bedret seg mye i løpet av de siste 30 årene, slik at majoriteten av barna kan regne med å bli helt friske (Forestier & Schmiegelow, 2006; G. Gustafsson et al., 2000; Kolmannskog et al., 2007; Pui, 2006; Pui & Evans, 2006; Pui, Schrappe, Ribeiro, & Niemeyer, 2004; Schmiegelow et al., 2010).
Behandling av leukemi hos barn er i Norge et regionalt ansvar. Barn med symptomer og funn som gir mistanke om leukemi, skal straks henvises tiltrengende øyeblikkelig hjelp til lokal og senere regional barneavdeling til utredning og behandling. Ofte må det startes væskebehandling og annen støttebehandling før overflytting til regionsykehus, se avsnittet om akutte onkologiske tilstander ovenfor. Når leukemidiagnosen er verifisert, startes behandlingen ved regionsykehusets barneavdeling, mens den videre behandlingen skjer i et nært samarbeid mellom regional og lokal barneavdeling.
Diagnose
Symptomer som kan gi mistanke om leukemi, er vanligvis uspesifikke: blekhet, blåmerker, hovne lymfeknuter, feber, skjelettsmerter. Ved klinisk undersøkelse finner man ofte at barnet ser påfallende sykt ut, og mange har hovne lymfeknuter, tegn på økt blødningstendens (blåmerker, petekkier) og forstørret lever og milt. Noen har hovne ledd. Klinisk undersøkelse må suppleres med blodprøver som ofte vil avsløre anemi, nøytropeni og trombocytopeni. Mange barn blir innlagt i barneavdeling pga. en akutt infeksjon uten at leukemimistanken er reist på forhånd.
Diagnosen stilles ved benmargsundersøkelse, kun unntaksvis kan diagnosen stilles ved mikroskopi av blodutstryk alene. Mikroskopi av benmargsutstryk må suppleres med benmargsbiopsi, immunfenotyping av benmargsaspirat, benmargskaryotyping og et sett molekylærbiologiske undersøkelser som er definert i behandlingsprotokollene. Det må også utføres spinalpunksjon for å se om det er spredning av leukemiceller til sentralnervesystemet (CNS), men denne prosedyren utsettes til diagnosen er sikker nok til at man kan gi første cellegiftdose rett inn i spinalvæsken samtidig.
Behandling
Behandling av leukemi hos barn skjer etter felles nordiske retningslinjer vedtatt av NOPHO (NOPHO – nordisk forening for pediatrisk hematologi og onkologi https://www.nopho.net/) som er en sammenslutning av nordiske barneonkologer fra alle de fem nordiske landene. NOPHO har egne behandlingsprotokoller for ALL og AML hos barn, og NOPHO har som gruppe tilsluttet seg Interfant-protokollen til behandling av ALL hos spedbarn, EsPhALL-protokollen til behandling av ALL med Philadelphia-kromosom, ICC APL Study 01 til behandling av akutt promyelocyttleukemi, ML-DS 2007 studien til behandling av barn med myeloid leukemi ved Down syndrom og EWOG-MDS 2006 til behandling av JMML hos barn. Alle protokollene er tilgjengelige på den lukkede delen av NOPHOs nettsider.
ALL
ALL deles inn i risikogrupper på bakgrunn av definerte risikofaktorer som er til stede ved diagnosetidspunktet, kombinert med respons på tidlig behandling. Risikofaktorene er modernisert og definert ut fra forskningsresultater oppnådd i NOPHOs tidligere behandlingsprotokoller (Forestier & Schmiegelow, 2006; G. Gustafsson et al., 2000; Kolmannskog et al., 2007; Schmiegelow et al., 2010).
Faktorer det tas hensyn til ved risikostratifisering av ALL:
- Antall hvite blodlegemer
- Alder
- Immunfenotype (B-precursor vs. T)
- Cytogenetikk – Visse definerte molekylærbiologiske karakteristika
- Respons på behandling definert som antall gjenværende leukemiske blaster i benmargen ved definerte tidspunkter.
I NOPHO ALL-2008 protokollen var alder tatt ut som selvstendig risikokriterium, dette er tatt inn igjen i den nye ALLTogether (A2G) protokollen. Generelt legges det nå mer vekt på cytogenetiske og molekylærbiologiske egenskaper ved de leukemiske blastene, kombinert med tidlig behandlingsrespons vurdert ved MRD (MRD – minimal residual disease) undersøkt ved hjelp av flowcytometri (pre-B-ALL) eller PCR-undersøkelser.
Den følgende risikostratifiseringen gjelder for A2G protokollen: Det skilles mellom Standard risk (SR), IR-low, IR-high, HR-chemo og HR-SCT. Tabellen nedenfor gir en oppsummering (tatt fra ALLTogether pilot versjon 1.4). Inndelingen er komplisert og stratifisering må i hvert tilfelle gjøres omhyggelig ved hjelp av nyeste protokollversjon.
| Stratification | Stratification criteria | |
| Standard-Risk (SR) | No T-cell ALL No HR-genetics* + No MRD-signal day 29 (end of induction) No CNS3/TLP+ No ABL-class fusion | |
| IR | MRD <5% day 29 | |
| IR-low (IRL) | <16 years at diagnosis No HR-genetics+MRD response as specified below | |
| Sub-group | MRD cut-off | |
| ETV/RUNX1 | MRD d29 <0.1% | |
| High hyperdiploid | MRD d29 <0.03% | |
| B-other + CNA GR** | MRD d29 <0.05% | |
| T-cell | No MRD signal d71 | |
| TLP+ <5 WBC/microL CSF | No MRD signal d29 | |
| IR-high (IRH) | Sub-group | MRD cut-off |
| CNS3-involvement TLP+ ≥5 WBC/microL CSF | Any MRD <HR-criteria | |
| Poorly responding testicular/mediastinal disease | Any MRD <HR-criteria | |
| CNA PR*** | Any detectable MRD d29 <5% | |
| Genetic groups as above | MRD ≥ cut-off above | |
| Failed genetic work-up | Any MRD <HR-criteria | |
| Failed MRD work-up | All patients | |
| T-cell | If MRD d29 <5% MRD <0.05% d 71 If MRD d29 ≥5% MRD <0.5% d 50 and undetectable d71 | |
| ≥ 16 years | Not SR/HR-criteria | |
| ABL-class fusions | Any MRD d29 MRD< 0.05% d71 | |
| HR-chemo BCP | t(17;19) and no HSCT-indication | |
| <16 years and MRD ≥5% d29 but MRD <0.05% d71 | ||
| <16 years, NCI HR at diagnosis and MRD ≥0.01% at TP2 | ||
| <16 years and remaining testicular disease/med mass ≥1/3 of initial volume after Consolidation 1 | ||
| HR-HSCT BCP | <16 years and MRD ≥0.05% d71 | |
| <16 years MRD d 29 ≥5% and ≥0.5% day 50 | ||
| ≥16 years and any HR-criteria | ||
| HR-HSCT T-cell | MRD d29 <5% and ≥0.05% day 71 | |
| <16 years MRD d 29 ≥5% and ≥0.5% day 50 | ||
| ≥ 16 years MRD ≥5% day 29 | ||
| <16 years MRD d29 ≥5%, <05% day 50 and detectable day 71 | ||
| Remaining testicular disease/med mass ≥1/3 of initial volume after Consolidation 1 | ||
Behandlingen i A2G-protokollen er intensiv kombinert cytostatikabehandling som bygger på tidligere NOPHO protokoller. Behandlingen er inndelt i induksjonsfase, konsolideringsfaser, sen intensivering og vedlikehold for SR- og IR-gruppene, mens pasienter med HR-ALL får, istedenfor en vanlig vedlikeholdsfase, intensivert kjemoterapi, stamcelletransplantasjon eller CAR-T. Varigheten av behandlingen er 2 år fra avsluttet induksjonsbehandling for alle risikogrupper unntatt pasienter som får behandling med SCT eller CAR-T.
Generelt om ALLTogether (A2G) protokollen
Protokollen er en kombinert behandlings- og forskningsprotokoll. Inklusjon krever skriftlig informert samtykke fra foreldrene. Barn som behandles etter denne protokollen, skal registreres online i ALLTogetherdatabasen. Registrering skal skje innen 1 uke etter diagnose og deretter fortløpende, hvor spesielt registrering av «events» (tilbakefall, manglende behandlingsrespons, død i komplett remisjon, sekundær kreft) og toksisitet er viktig.
- Induksjonsfasen består av dexamethason, vinkristin, asparaginase, og intratekal behandling (medikamenter som settes direkte inn i spinalvæsken for å beskytte mot leukemi i CNS). Pasienter med T-celle ALL og andre risikokriterier får i tillegg daunorubicin.
- Første konsolideringsfasen er avhengig av risikogruppe (SR vs. høyere risiko) og består av nedtrappende dexamethason etterfulgt av peroral 6-merkaptopurin og 4 uker med lavdose cytarabin samt 2–3 doser asparaginase. I tillegg får de høyere risikogruppene cyklofosfamid og intratekal behandling
- Andre konsolideringsfaser er avhengige av risikogruppe og består bl.a. av asparaginase og høydose-metotrexat kurer. Antallet HD-MTX kurer er dog vesentlig redusert i denne protokollen sammenlignet med tidligere NOPHO protokoller. Foreløpig (i pilotfasen) får alle pasienter doxorubicin i sen-intensiveringen, senere vil SR pasienter som randomiseres til eksperimentell arm kunne få hele behandlingen uten doxorubicin og dermed uten antracykliner i det hele tatt.
- Vedlikeholdsbehandlingen inneholder 6-merkaptopurin og metotreksat i tablett- eller miksturform og for alle risikogrupper unntatt SR gjentatte pulser med vincristin/dexamethason.
Protokollen inneholder i pilotfasen ingen randomiseringer. Når master-protokollen åpner i løpet av 2020 vil det være 3 randomiseringer:
- Randomisering 1: Standard riskgruppe. I senintensivering randomiseres pasienter mellom doxorubicin (standard arm) og ikke doxorubicin (eksperimentell arm), kfr. ovenfor. Dette er en non-inferiority studie.
- Randomisering 2: Intermediate-low riskgruppe. Randomisering i 3 grupper: Standard arm: doxorubicin i senintensivering og vincristin/dexapulser i vedlikeholdsfasen. Eksperimentell arm 1: Ikke doxorubicin i senintensivering men vincristin/dexapulser i vedlikehold. Eksperimentell arm 2: Doxorubicin i senintensivering men ikke vincristin/dexapulser i vedlikehold.
- Randomisering 3: Intermediate-high riskgruppe. Randomisering av vedlikeholdsbehandlingen i 3 grupper: Standard arm: Vincristin/Dexa pulser hele veien til behandlingen avsluttes. Eksperimentell arm 1: Inotuzumab (eksperimentell terapi) i 6 uker, deretter VCR-dexa pulser som i standard arm. Eksperimentell arm 2: Som standard arm, men tillegg av 6-tioguanin (TEAM).
For hver randomisering kreves eget samtykke fra foreldrene.
Anbefalinger:
- Pasienter med ALL bør behandles etter den til enhver tid gjeldende NOPHO-protokollen, p.t. er dette A2G protokollen (pilot versjon) mens fullversjonen av A2G forventes åpnet i løpet av 2020. Det kreves eget samtykke fra foreldre.
- Et fåtall pasienter behandles med stamcelletransplantasjon eller CAR-T i første remisjon.
Spedbarns-ALL
Hos barn som er < 1 år ved diagnose, anbefaler NOPHO bruk av «Interfant 06»-protokollen, en videreutvikling av Interfant-99 protokollen, som er hittil beste publiserte behandling (Pieters et al., 2007) (Evidensgrad B). Protokollen ligner på intermediærrisikogruppen av den ordinære ALL-protokollen, men inneholder mer cytarabin, noe som anses som gunstig ved MLL-rearrangeringer som er til stede i de fleste tilfellene av spedbarns-ALL. Protokollen inneholdt tidligere en randomisering, hvor den eksperimentelle armen hadde 2 AML-lignende blokker, dette er avsluttet og man bruker standard arm. Protokollen brukes i Norge som «best available treatment», og man har fulgt standard arm hele tiden. Alle risikogrupper følger prinsipielt samme behandling, men pasienter i høyrisikogruppen og pasienter i intermediærgruppen med dårlig terapirespons tilbys i tillegg stamcelletransplantasjon (G. Mann et al., 2010) (Evidensgrad B).
Anbefalinger:
- Spedbarn med ALL bør behandles etter Interfant 06-protokollen, og følge standardarm. Det kreves eget samtykke fra foreldre. (Evidensgrad B)
- Pasienter i høyrisikogruppen og pasienter i intermediærgruppen med dårlig respons (definisjon, se protokoll) tilbys stamcelletransplantasjon. (Evidensgrad B)
Philadelphia-kromosom-positiv ALL
Ca 3 % av barn med ALL får påvist Philadelphia-kromosom (bcr-abl) i forbindelse med utredning for ALL. Siden dette svaret ikke er kjent fra starten, vil barna starte behandling etter NOPHO ALL 2008 ( evt A2G)-protokollen, men etter induksjonsfasen overføres de til behandling etter en internasjonal protokoll, EsPhALL, tilpasset nordiske forhold av NOPHO («EsPhALL amendement for NOPHO»). Det som skiller behandlingen av barn etter denne protokoll fra ALL-behandlingen for øvrig, er først og fremst behandling med imatinib (Glivec®) som gis fra dag 15 og videre under hele behandlingsforløpet. Også etter eventuell stamcelletransplantasjon gis imatinib i ett år. Under behandlingen monitoreres tilstedeværelsen av bcr-abl-translokasjonen regelmessig, og resultatene av denne monitoreringen vil avgjøre om pasienten er kandidat for stamcelletransplantasjon eller ikke. Det må understrekes at indikasjonen for stamcelletransplantasjon er under kontinuerlig revurdering. En ny protokoll for Ph+ALL er på vei, men foreløpig ikke godkjent i Norge.
Anbefalinger:
- Ph+ ALL pasienter bør behandles etter EsPhALL-protokollen («EsPhALL amendement for NOPHO») med imatinib og eventuelt stamcelletransplantasjon. Det kreves eget samtykke fra foreldre. (Evidensgrad B)
ALL-residiv
Ca 15 % av barn behandlet for ALL vil senere få residiv. Siden ALL er den hyppigste barnekreftformen, vil det si at ALL-residiv kommer opp i et antall av 5–10 nye tilfeller i Norge årlig. Prognosen etter residiv av ALL er dårligere enn ved første gangs behandling, med opp mot 50 % sjanse til helbredelse ved sene residiver, dårligere ved tidlige residiver. Hvis residivet opptrer tidlig (mindre enn 6 måneder etter avsluttet primærbehandling – høy risiko), vil pasienten tilbys stamcelletransplantasjon etter oppnådd ny remisjon. Ved sene residiver (standard risiko) er det mest vanlig å behandle med kjemoterapi alene, men avhengig av terapirespons vurdert ved MRD, vil det også her kunne bli aktuelt med stamcelletransplantasjon.
Det har tradisjonelt ikke vært noen ensartet behandling av residiv i Norge eller Norden. Mest brukt har den tyske ALL-REZ BFM 2002-protokollen vært, i noen grad også den engelske ALL UKR3-protokollen. Pasienter med residiv av standard risiko behandles nå etter IntReALL-SR-2010-protokollen som ble åpnet i 2014. Denne protokollen blir prinsippene i den tyske og den engelske tilnærmingen sammenlignet. Norge er tilsluttet denne protokollen. I tillegg er det åpnet en høy risiko del av denne protokollen. Det anbefales å rådføre seg med nasjonal ALL koordinator ved alle nydiagnostiserte residiver.
AML
De Nordiske landene har tradisjonelt sett hatt svært gode resultater i behandling av AML hos barn (Hasle et al., 2012; Lie et al., 2005). AML behandles i henhold til NOPHO-DBH-AML 2012-protokollen. Cytostatikabehandlingen er basert på en kombinasjon av antracykliner, cytarabin og etoposid i induksjonsfasen (første 2 kurer), samt høydose cytarabin alene eller kombinert med etoposid eller anthracyklin i konsolideringskurene. Måling av MRD (minimal restsykdom) ved flowcytometri (evt. ved PCR dersom det ikke er mulig å følge MRD via flow) er kommet inn som stratifiserende faktor i den nye protokollen. Pasientene klassifiseres som standard risiko (SR) eller høy-risiko (HR) basert på behandlingsrespons på de første 2 kurene. Pasienter med > 15 % blaster etter 1. kur samt pasienter med intermediær respons (0,1 til 4,9 % blaster etter kur 2) stratifiseres til HR gruppen. Pasienter med ≥ 5 % blaster etter kur 2 går ut av protokollen og behandles individuelt med ett av flere mulige salvage-regimer. Også pasienter med FLT3 ITD mutasjon (med NPM1 wild type) stratifiseres som HR. SR-gruppen behandles med totalt 5 svært intensive cytostatikakurer. HR-gruppen tilbys stamcelletranplantasjon i første remisjon, vanligvis etter 3. eller 4. kur.
Protokollen inneholdt fra starten av 2 randomiseringer. I første induksjonskur ble anthracyklinene mitoxantron og liposomalt daunorubicin sammenlignet med hverandre. Denne randomiseringen er nå avsluttet og anbefalingen er å bruke MEC (kuren som inneholder Mitoxantron) hos alle pasienter, da denne kuren har gitt signifikant forbedret overlevelse. I andre induksjonskur randomiseres fortsatt mellom kur ADxE (cytarabin, liposomalt daunorubicin, etoposid) og kur FLADx (fludarabin, cytarabin, liposomalt daunorubicin). Pga verdensomspennende mangel på Daunoxome, kan Daunoxome erstattes med Daunorubicin (mens Daunoxome ikke er tilgjengelig). Den nordiske protokollen inneholder høye kumulative antracyklindoser, og regelmessige ekkokardiografiske kontroller som spesifisert i protokollen er svært viktig (Lie et al., 2005) (Evidensgrad C).
En undergruppe av AML kjennetegnes ved t(15;17) (PML-RARA) i de leukemiske blastene (akutt promyelocyttleukemi – APL, FAB-klassse M3). Disse pasientene har ofte en uttalt koagulopati på diagnosetidspunktet, som ved DIC, og de krever umiddelbar behandling med «all-trans-retinoic acid» – ATRA, som raskt vil bedre blødningstilstanden. Pasienter med APL har etter et vedtak i NOPHO blitt behandlet etter protokoll «ICC APL Study 01», men en ny protokoll for SR APL er under utarbeidelse (ICC APL study 02). Den nye protokollen anbefales brukt allerede nå (se anbefaling på https://www.nopho.net/ under protokoller/APL). Denne nye protokollen kjennetegnes ved bruk av arsen-trioxyd (ATO) i tillegg til høydosert vitamin A (all-trans-retinoic acid, ATRA, = Vesanoid®) og inneholder ikke lenger vanlige cytostatikakurer. PCR-baserte MRD-undersøkelser underveis i behandlingen er svært viktige, da det finnes en effektiv residivbehandling som er skissert i samme protokollen. Akutt promylocyttleukemi er så ekstremt sjeldent i Norge (< 1 tilfelle per år hos barn) at man bør ta kontakt med nasjonal AML koordinator/NOPHO AML gruppen dersom et tilfelle er diagnostisert eller mistenkt.
Anbefalinger:
- Pasienter med AML bør behandles etter den til enhver tid gjeldende NOPHO-protokollen, for tiden NOPHO-DBH-AML 2012 protokollen. Det kreves eget samtykke fra foreldre.
- Pasienter i høyrisikogruppen (inkl. pasienter med FLT3 ITD og samtidig NPM1 wild type) bør tilbys allogen stamcelletransplantasjon i første remisjon.
- Regelmessige ekkokardiografiske kontroller er spesifisert i protokollen som svært viktig.
- Undergruppen av AML (FAB M3, PML-RARA) skal behandles etter internasjonal APL protokoll med med modifikasjoner – ta kontakt med NOPHO-AML gruppen.
Myeloid leukemi ved Downs syndrom
Dette er en egen leukemiform som bare finnes hos barn med Downs syndrom, og da bare i alderen 0–4 år. I blastene påvises alltid GATA-1 mutasjonen. Mange av barna har gjennomgått transitorisk abnorm myelopoiese (TAM) i nyfødtperioden, og av Down-barn som har TAM i nyfødtperioden, vil ca. 25–30 % få AML senere. Diagnosen forutgås ofte av en periode med cytopeni, mest typisk trombocytopeni.
Barn med Down syndrom har nedsatt toleranse for en del cytostatika, men det er dokumentert at de har svært god prognose med en overlevelse opp mot 90 %, selv om de får redusert behandling i forhold til andre barn med AML (Abildgaard et al., 2006; Creutzig et al., 2005; Zeller et al., 2005) (Evidensgrad C). Av den grunn har NOPHO anbefalt å følge den europeiske protokollen «ML-DS2006». Behandlingen i denne protokollen er basert på konvensjonell AML-behandling, men inneholder mindre anthracykliner, og det gis kun 4 kurer.
Anbefalinger:
- Den europeiske ML-DS 2006-protokollen anbefales brukt (Evidensgrad C).
Kronisk Myelogen Leukemi (KML)
KML hos barn er sjelden, men forekommer. Barna behandles etter samme retningslinjer som de voksne med imatinib, men det er foreløpig uavklart om de kan klare seg uten stamcelletransplantasjon på lang sikt. Ved nedsatt respons på imatinib, primært eller sekundært, kan man forsøke annen-generasjons tyrosin kinasehemmer som brukes hos voksne (nilotinib eller dasatinib) (Andolina, Neudorf, & Corey, 2012). Det arbeides med å opprette et internasjonalt register for barn med KML.
Anbefalinger:
- KML hos barn behandles med imatinib som hos voksne.
- Som andrelinjebehandling anbefales nilotinib eller dasatinib.
- Det er foreløpig uavklart hvilken plass stamcelletransplantasjon vil komme til å få i behandlingen i fremtiden.
Juvenil myelomononcyttleukemi (JMML)
En sykdom som rammer fortrinnsvis små barn. Utredningen er detaljert beskrevet i protokollen «EWOG-MDS 2006». Ved mistanke om JMML skal spesialprøver sendes til laboratoriet i Freiburg, Tyskland, rekvisjoner og adresser finnes i protokollen. Barn med JMML kan bare helbredes med hematopoietisk stamcelletranplantasjon. Man prøver å unngå å gi kjemoterapi før transplantasjonen, men noen ganger er dette nødvendig pga svært høye celletall, respirasjonsproblemer eller andre komplikasjoner (B. Gustafsson, Hellebostad, Ifversen, Sander, & Hasle, 2011) (Evidensgrad C). Graft-versus-leukemi-effekten er svært viktig ved JMML, og etter stamcelletransplantasjonen er det derfor nødvendig å redusere og seponere immunsuppresjonen tidlig, helst innen 2–3 måneder. Tilstanden kan forekomme forbigående hos pasienter med «Noonan-syndrom» og løser seg da spontant. Ved JMML anbefales å ta kontakt med nasjonal og/eller nordisk koordinator for EWOG-MDS tidlig.
Anbefalinger:
- JMML pasienter bør behandles etter EWOG-MDS 2006-protokollen.
- Ved JMML er hematopoietisk stamcelletransplantasjon eneste kurative terapi. Man prøver å unngå kjemoterapi før transplantasjonen (Evidensgrad C). Etter stamcelletransplantasjonen bør immunsuppressiv behandling helst seponeres innen 3 måneder.
Non-Hodgkin lymfom
Non-Hodgkin lymfom (NHL) hos barn og ungdom er en heterogen gruppe av maligne tilstander med opphav i det lymfatiske systemet. Sykdomsspekteret varierer fra transformasjon av umodne T- eller B-celleforløpere (lymfoblastiske lymfomer) til neoplasmer av modne, differensierte lymfocytter (perifere T- eller B- celle NHL) (Reiter, 2007). Forekomst av de ulike NHL-subtypene i barne- og ungdomsalderen avviker fra fordelingen hos voksne pasienter.
Hos barn dominerer følgende typer:
- lymfoblastiske lymfomer (ca. 25 % av alle NHL hos barn)
a) med subtyper B- og T-lymfoblastisk lymfom - perifere B-celle NHL (ca. 55 %)
a) med dominans av Burkitt lymfom (ca. 60 % av B-NHL), ellers
b) diffust stor-cellet B-celle lymfom (DLBCL)
c) moden B-celle leukemi (B-ALL)
d) primært mediastinalt B-celle lymfom - perifere T-celle NHL (ca. 15 %)
a) inkludert anaplastisk stor-cellet lymfom (ALCL) (> 90 % av T-NHL).
Alle andre subtyper er svært sjeldne eller opptrer ikke hos barn. Årlig insidens i Norge er ca. 5 pasienter i aldersgruppen 0–14 år (ca. 6 % av alle malignesykdommer hos barn).
Diagnose
Non-Hodgkin lymfom er kjennetegnet av ukontrollert vekst i lymfevev. Hovedsymptom ved NHL er uømme, forstørrede lymfeknuter ved nodal sykdom, ellers en rekke ulike symptomer ved ekstranodale manifestasjoner. Symptomene varierer med lokalisasjon og utbredelse (f.eks. magesmerter ved abdominelt NHL, åndenød ved mediastinalt lymfom). Hyppigste lokalisasjoner er cervikale lymfeknuter, abdomen, mediastinum og ØNH-området, for øvrig bein, beinmarg, CNS, epiduralrom, nyre, testikler, ovarier, hud og bløtdeler. Allmennsymptomer som nattesvette, vekttap, tretthet og feber kan forekomme. Noen pasienter er i nesten upåvirket allmenntilstand, mens andre presenterer seg med livstruende komplikasjoner (f.eks. trakeakompresjon, tverrsnittslesjoner og nyresvikt).
Diagnostikken ved NHL er omfattende og baseres på radiologiske, histopatologiske, immunfenotypiske og i økende grad cytogenetiske og molekylære undersøkelser. Eksakt klassifisering er en forutsetning for å kunne velge korrekt behandlingsprotokoll.
Biopsi av tumormaterialet må være av tilstrekkelig kvalitet og kvantitet, helst en hel lymfeknute dersom dette er mulig uten betydelig risiko. Tumormaterialet sendes til histologi, immunfenotyping (immunhistokjemi, flowcytometri), cytogenetikk (NHL-spesifikke kromosomtranslokasjoner) og molekylærgenetikk (NHL-spesifikke genmutasjoner).
I noen tilfeller vil man kunne stille diagnosen uten operasjon (biopsi), f.eks. ved cytologisk og immunfenotypisk analyse av blod, beinmarg eller pleuravæske.
Obligate undersøkelser i forbindelse med stadieinndelingen inkluderer ultralyd av abdomen, klinisk undersøkelse av testikler (og evt ultralyd), rtg thorax, MR (CT) av alle regioner med forstørrede lymfeknuter, ved CNS symptomer MR caput, MR spinalkanal ved neurologiske utfall, spinalpunksjon, benmargsundersøkelser og ved tidligere hjertesykdom eller symptomer på hjertesykdom ekko cor og EKG. Verdien av PET/CT i NHL-diagnostikken og oppfølgingen er ikke entydig avklart og vil derfor bli evaluert i kommende NHL-studier. Per februar 2019 inngår ikke PET/CT rutinemessig i utredningen av NHL hos barn.
Anbefalinger:
- Obligate undersøkelser i forbindelse med stadieinndelingen inkluderer ultralyd av abdomen, klinisk undersøkelse av testikler (og vt ultralyd), rtg thorax, MR (CT) av alle regioner med forstørrede lymfeknuter, MR caput / spinalkanal ved neurologiske utfall, spinalpunksjon, benmargsundersøkelser, måling av laktat dehydrgenase (LDH) og eventuelt ekko cor og EKG.
Stadieinndeling
Stadium I | En enkel nodal eller ekstranodal tumormanifestasjon uten lokal spredning, bortsett fra mediastinale, abdominale eller CNS/benmargsengasjement. |
Stadium II | En singel ekstranodal manifestasjon med lokal lymfeknutespredning. To eller flere nodale og eller ekstranodale manifestasjoner på samme side av diafragma, med eller uten lokal spredning, bortsett fra mediastinale, epidurale eller utbredte, ikke receserbare abdominale lokalisasjoner, eller benmargsengasjement. |
Stadium III | Lokalisasjoner på begge sider av diafragma. |
Stadium IV | Beinmarg- og/eller CNS-affeksjon (uansett annen lokalisasjon i tillegg). |
Risikostratifisering
B-NHL og ALCL deles inn i risikogrupper. Risikogruppen avgjør behandlingsintensiteten.
B-cellelymfom (iht protokoll B-NHL 2013)
Risikogruppe 1
Makroskopisk fullstendig fjernet tumormanifestasjon
Risikogruppe 2, st I/II
Inkomplett reseksjon. Stadium I + II sykdom
Risikogruppe 2, st III
Inkomplett reseksjon. Stadium III + LDH < 2 x øvre referanseområde
Risikogruppe 3
Inkomplett reseksjon. Stadium III med LD >= 2x øvre referanseområde men < 4x øvre referanseområde, samt stadium IV/B-ALL med LD < 1000 U/l og ingen CNS-sykdom
Risikogruppe 4
Inkomplett reseksjon. Stadium III med LD >=4 x øvre referanseområde, samt stadium IV/B-ALL med LD > = 4x øvrereferanseområde uten CNS-sykdom
Risikogruppe 4, CNS +
Inkomplett reseksjon. Stadium IV/B-ALL inkludert CNS engasjement
Storcellet anaplastisk lymfom (iht protokoll ALCL 99)
Lav-risikogruppe
Sykdom i stadium I som har blitt komplett recesert Standard-risikogruppe.
Inkomplett reseksjon. Ingen hudaffeksjon, ingen mediastinaltumor, ingen affeksjon av lever, milt eller lunge.
Høy-risikogruppe
Inkluderer pasienter med en av følgende:
Hudlesjoner (biopsi), mediastinaltumor, lever-, milt- eller lungeaffeksjon
Behandling
Primærutredningen og ansvaret for behandling av Non-Hodgkin lymfom hos barn og ungdom tilligger de fire barneonkologiske sentraene ved universitetsykehusene. Alle de nordiske land følger samme behandlingsprotokoller i samråd med NOPHO (Nordic Society of Paediatric Haematology and Oncology).
Behandlingsstrategi: lymfoblastiske lymfomer behandles i hht strategien for akutt lymfatisk leukemi (ALL) med tilstrebet kontinuerlig cytostatika kombinasjonsbehandling over en lang periode (18–24 mnd). Ikke-lymfoblastiske lymfomer behandles med kortere, intensive kombinasjonskurer med høy doseintensitet. Ti års overlevelse for hele gruppen av Non-Hodgkin lymfom hos barn er 87–93 % (Nasjonalt kvalitetsregister for barnekreft, 2018) .
Etter internasjonal konsensus deles lymfombehandlingen hos barn inn i 3 terapeutiske hovedgrupper:
- Lymfoblastisk lymfom: I Norge behandles både T- og B-lymfoblastisk lymfom etter den europeiske behandlingsprotokollen LBL 2018. Protokollen er basert på den tidligere behandlingsprotokollen Euro-LB 02. Protokollen er per våren 2019 åpen for inklusjon av norske pasienter. Protokollen inkluderer induksjons-, konsoliderings- og vedlikeholdsbehandling med systemisk og intratekal cytostatika kombinasjonsbehandling som inkluderer vinkristin, prednisolon, deksametason, methotrexat, daunorubicin, doksorubicin, asparaginase, cyklofosfamid, cytarabin, 6-mercaptopurin og 6-thioguanin. Behandlingsintensiteten tilpasses sykdomsutbredelsen (stadium). «Event-free survival» for hele gruppen er ca. 80–90 %.
- Perifere B-NHL: Norge deltar i studieprotokollen B-NHL 2013, som har vært åpen i Norge siden 2018. I protokollen inngår 2 randomiseringer (Norge deltar per februar 2020 kun i en av randomiseringene), og bruk av Rituximab og undersøkelse av immunrekonstitusjon etter Rituximab er en av hovedmålene med studien. Det er fire ulike risikogrupper (RI-RIV) og per februar 2019 får alle pasientene Riuximab som en del av behandlingen. Behandlingen for øvrig inkluderer (i ulik grad avhengig av risikogruppe) vinkristin, vindesin, deksametason, prednisolon, methotrexat, doksorubicin, etoposid, cyklofosfamid, ifosfamid og cytarabin. CNS-positive pasienter får intensivert intrathekal behandling. Stråling inngår ikke i behandlingen. «Event-free survival» for hele gruppen er ca. 85–90 %.
Rituximab er et monoklonalt antistoff rettet mot overflateantigenet CD20 på lymfatiske B‑celler og lymfomceller. To prospektive kliniske studier har de siste årene vist at infusjon av Rituximab har antitumoraktivitet mot modne B- cellelymfomer hos barn – både dersom gitt som eneste medikament og additivt i kombinasjon med standard kjemoterapibehandling (Goldman et al., 2013; Meinhardt et al., 2010). I aktuell behandlingsprotokoll anser man at Rituximab har en viktig rolle i behandlingen av avansert modent B-cellelymfom hos barn.
- Anaplastisk stor-cellet lymfom (ALCL): I Norge behandles ALCL hos barn etter den internasjonale protokollen ALCL99 (pr. feb. 2020). Standardarmen i protokollen (ingen randomisering) brukes nå som «best available treatment». Kombinert intratekal og systemisk cytostatikabehandling som inkluderer deksametason, cyklofosfamid, cytarabin, ifosfamid, methotrexat, etoposide og doksorubicin, ingen randomisering. Behandlingsintensiteten tilpasses sykdomsutbredelsen (stadium). Stråling inngår ikke. «Event-free survival» for hele gruppen er ca. 70 %. En ny europeisk behandlingsprotokoll for ALCL er i avsluttende planleggingsfase, det er usikkert om Norge vil kunne delta aktivt i denne.
Anbefalinger:
- Pasienter med lymfom bør behandles etter den til enhver tid gjeldende behandlingsprotokoll anbefalt av NOPHO-lymfomgruppen for hver av de terapeutiske lymfom- hovedgrupper. Det kreves samtykke fra foreldrene for inklusjon i de aktuelle protokollstudiene og evt i NOPHO registeret.
- For svært sjeldne lymfomgrupper og ved residiv av NHL må behandlingen individualiseres, etter nasjonal og evt internasjonal diskusjon.
Behandling ved residiv
Tidsperioden med størst risiko for tilbakefall varierer med NHL-subtype og er for B-NHL ca. 6–9 mnd fra diagnosetidspunktet, for ALCL ca. 15 mnd. T- lymfoblastisk lymfom (T-LBL) kan residivere svært tidlig under pågående eller rett etter avsluttet behandling, mens residiver av B-LBL kan også opptre etter noen år.
Residiver ved lymfoblastisk lymfom blir ofte behandlet etter protokoller for ALL- residiv, og strategien inkluder allogen stamcelletransplantasjon. Likevel er langtidsprognosen per i dag svært dårlig (toksisitet, progress). Det samme gjelder for residiv ved moden B-NHL (som Burkitt), til tross for behandling med høyintensive cytostatikakombinasjoner og allogen (eller autolog) transplantasjon. Ved moden B-NHL residiv er tidspunktet for transplantasjon kritisk (dvs straks etter oppnådd remisjon), type transplantasjon (allogen vs autolog) er sannsynligvis av mindre betydning for overlevelsen (Minard-Colin et al., 2015). For CNS-tilbakefall av B-NHL er det rapporter som tyder på at Rituximab trygt og effektivt kan gis intraventrikulært/intrathekalt (Ceppi et al., 2016). Prognosen ved ALCL residiv er klart bedre enn ved andre NHL-subtyper, og kan ofte kureres ved monoterapi med Vinblastin, men i noen tilfeller er det nødvendig med allogen stamcelletransplantasjon og i motsetning til andre B-NHL residiv er det ikke like viktig med fullstendig sykdomsremisjon før transplantasjon. ALCL residiv behandles etter ALCL relapse 04 (med amendment 2013). Det er kommet en del nyere medikamenter som har vært brukt i ulike NHL residiv sammenhenger (ALK hemmere, Brentuximab Vedotin, Blinatumomab, CAR-T m.fl). Disse vil bli vurdert i hvert enkelt tilfelle av residiv og det foreligger ingen standard indikasjon for disse.
Kontrollopplegg for Non-Hodgkin lymfom
Avhengig av NHL-subtype. Hver protokoll inneholder som oftest detaljert beskrivelse av kontrollenes hyppighet og omfang av diagnostikk. Orienterende og veiledende gjelder:
Billeddiagnostikk av affisert område (helst ultralyd, men avhengig av lokalisasjon evt. MR, CT, rtg):
- Lymfoblastisk lymfom: Residiverer tidlig, derfor rtg thorax hver 3. mnd. i vedlikeholdsfasen; i 1. året etter behandling billeddiagnostikk hver 3. mnd, i 2. året hver 6. mnd, deretter kun ved mistanke om tilbakefall. BM og CNS-diagnostikk kun ved mistanke om residiv.
- B-NHL: hver 3–6. mnd etter avsluttet terapi første 18 mnd (avhengig av protokoll); deretter hver 6. mnd frem til (2-)3 år etter behandlingsslutt. Deretter kun ved mistanke om tilbakefall. BM og CNS-diagnostikk også kun ved mistanke om residiv.
- ALCL: hver 3. mnd etter avsluttet terapi første året, hver 4. mnd andre året, hver 6. mnd i totalt 5 år.
Klinisk undersøkelse og blodprøver:
- Hver 1–3 mnd. første år etter avsluttet terapi, deretter hver 3.–6. mnd, avhengig av protokoll.
Ekkokardiografi/nyrefunksjon/gonadefunksjon:
- Avhengig av behandling.
Hodgkin lymfom
Hodgkins lymfom (HL) er et malignt lymfom utgående fra B-lymfocytter. HL består av to entiteter; klassisk HL (med undergruppene lymfocyttrik type, nodulær sklerose, bladet cellularitet og lymfocyttfattig type) og nodulært lymfocyttrikt HL (tidligere paragranulom). Klassisk HL karakteriseres histologisk av et lite antall tumorceller [Hodgkin/Reed-Sternberg (HRS)–celler] i et vev dominert av ikke neoplastiske celler. Ved nodulær lymfocytt rikt HL finnes de såkalte «lymphocyte predominant» (LP)-celler. Det diagnostiseres ca. 140 nye tilfeller av HL pr. år i Norge (Larsen, 2018). En insidenstopp observeres hos unge voksne i aldersgruppen 15–34 år, en annen hos eldre over 50 år. Det er ca. 10 tilfeller hos barn og ungdom under 18 år årlig. Klassisk HL utgjør ca. 95 % av tilfellene og nodulært lymfocyttrikt HL 5 %.
Overlevelsen ved HL har bedret seg betydelig gjennom de siste 50 år. Dette skyldes effektive cytostatikaregimer og bedret bruk av strålebehandling. Siden 1998 har Norge deltatt i studier i regi av Deutsche Arbeitsgemeinschaft für Leukemie (DAL), senere Gesellschaft für Pädiatrische Onkologie und Hämatologie (GPOH). Fra 2008 er dette videreutviklet til et europeisk samarbeid, EURONET, der Norge deltar. Med disse protokollene er prognosen for barn og ungdom med HL generelt svært god.
Diagnose
Klassiske symptomer ved HL er glandelsvulst og vekttap/fravær av adekvat vektøking for alderen, feber, patologisk nattesvette (såkalte B-symptomer) eller andre allmennsymptomer, f.eks hudkløe. Ved undersøkelse legges spesielt vekt på patologisk glandelsvulst og hepato-splenomegali. Blodprøver er ikke spesifikke for HL, men viser ofte bl.a. høy SR, leukocytose med granulocytose, lymfopeni, anemi, trombocytose, høy CRP og gjerne lav albumin. Diagnosen stilles ved en skjærebiopsi av affisert lymfeknute eller tilsvarende. Det er viktig å være klar over at finnålsaspirasjons-cytologi alene kan være falsk negativ ved mange lymfomer, spesielt ved HL, og at cytologi aldri alene er godt nok for å diagnostisere HL. For histopatologisk undersøkelse, se Nasjonalt handlingsprogram med retningslinjer for diagnostikk, behandling og oppfølging av maligne lymfomer (Helsedirektoratet, 2019).
Utbredelsesdiagnostikk ved HL inkluderer minimum en full anamnese med blant annet fokus på B-symptomer, klinisk undersøkelse, bredt anlagte blodprøver (inkludert senkningsreaksjon (SR)), ØNH-undersøkelse (kan utelates), røntgen thorax (kan utelates), ultralydundersøkelser av abdomen, MR av hals, thorax, abdomen og bekken samt CT av thorax. PET/CT er en standard modalitet for diagnostikk og måling av behandlingseffekt. PET-CT vurdert av trenede nuklærmedisinere kan erstatte benmargsbiopsi, som ellers skal tas. (for detaljer se Nasjonalt handlingsprogram med retningslinjer for diagnostikk, behandling og oppfølging av maligne lymfomer (Helsedirektoratet, 2019).
Ann Arbor klassifikasjonen brukes for stadieinndeling av HL og danner basis for behandlingsgrupper (se nedenfor).
Stadium I | Sykdom i en lymfeknuteregion (milt, thymus og Waldeyers ring regnes som nodal affeksjon). |
Stadium II | Sykdom i to eller flere lymfeknuteregioner på samme side av diafragma eller en eller flere lymfeknuteregioner på samme side av diafragma med innvekst i ekstralymfatisk organ/vev (IIE). |
Stadium III | Sykdom i lymfeknuteregioner på begge sider av diafragma eller en eller flere lymfeknuteregioner på begge sider av diafragma med innvekst i ekstralymfatisk organ/vev (IIIE). |
Stadium IV | Diffus eller disseminert sykdom i et eller flere ekstralymfatiske organ/vev med eller uten affeksjon av lymfeknuter. |
Tilleggsopplysninger til Ann Arbor
- Ekstranodal vekst: Markeres med suffikset E. Lokalisert affeksjon av vev/organ i tilslutning til affiserte lymfeknuter slik at direkte innvekst vekst kan antas.
- Bulky sykdom: Markeres med suffikset X (definisjonen varierer etter protokoll).
- B-symptomer: Inndeling i undergruppene A eller B avhengig av om pasienten ikke har eller har ett eller flere B symptomer:
- Uforklarlig vekttap på mer enn 10 % siste 6 måneder
- Uforklarlig persisterende eller residiverende feber med temperatur over 38 °C siste måned
- Gjentatt kraftig nattesvette siste måned.
Risikostratifisering
I den pågående Euronet –PHL-C2 studien defineres 3 terapinivåer (treatment level, TL), basert i hovedsak på stadieinndeling etter Ann Arbor klassifikasjonen. Det skilles mellom 3 nivåer:
TL-1 | Stadium IA/IB/IIA med SR < 30 mm/h og fravær av bulky sykdom her definert som største lesjon over 200 ml. |
TL-2 | Stadium IA/IB/IIA med SR >30 mm/h og/eller bulky sykdom over 200 ml, stadium IAE/IBE/IIAE/IIB/IIIA |
TL-3 | Stadium IIBE/IIIAE/IIIB/IV |
Anbefalinger diagnostikk:
- Diagnosen stilles ved en skjærebiopsi av affisert lymfeknute eller tilsvarende.
- Utbredelsesdiagnostikk som anført.
- PET/CT er vesentlig før, under og etter behandling.
- Benmargsbiopsi kan utelates dersom PET-CT evaluerer benmargsaffeksjon.
Hodgkins lymfom – behandling
Ansvaret for behandling av HL tilligger universitetsykehusene. Behandling ved nodulært lymfocyttrikt HL og klassisk HL er noe forskjellig, og de omtales derfor hver for seg.
Behandling ved nodulært lymfocyttrikt Hodgkins lymfom
Denne subgruppen av HL presenterer seg regelmessig i stadium I eller II. Ikke sjelden er all synlig tumor fjernet ved kirurgi for biopsitaking. Data taler for at slike pasienter kan observeres uten videre behandling (Mauz-Korholz et al., 2007) (evidensgrad C). De fleste residiver etter kirurgi alene vil være lokaliserte og ikke føre til et høyere stadium eller mer komplisert behandling for pasientene enn om de fikk onkologisk tilleggsbehandling første gang. En europeisk studie, Euronet-PHL-LP1, startet i 2010 for blant annet prospektivt å evaluere denne tilnærmingen ved kirurgisk resesert sykdom. Norge deltar ikke i denne studien per i dag. For pasienter med restsykdom etter kirurgi, residiv etter komplett kirurgisk reseksjon eller utbredt sykdom blir behandlingen lik den som gis for tilsvarende stadier av klassisk HL.
Sene residiver etter kjemoterapi og strålebehandling forekommer ikke sjelden, og de har ofte et indolent preg. Standard terapi baseres på samme prinsipper som ved klassisk HL, selv om det mer usikkert om man kan oppnå varig kurativ effekt. Nytten av HMAS er heller ikke godt dokumentert. Regimer som IEP, ABVD, LVPP eller MIME kan forsøkes. Det kan være aktuelt å gi disse regimene kombinert med rituximab siden lymfomet er CD20 positivt. Rituximab monoterapi kan forsøkes idet det er dokumentert responsrater hos voksne (Schulz et al., 2008). Stråleterapi mot problemområder forventes å ha god effekt.
Anbefalinger:
- Kirurgi anbefales når tumorreseksjon er mulig (Evidensgrad C).
- Ved residiver/ikke resesert sykdom gis tilsvarende kjemoterapi som ved klassisk Hodgkins lymfom (se nedenfor).
- Ved sene residiver kan det være aktuelt å prøve rituximab monoterapi eller som kombinasjonsbehandling (Evidensgrad C).
Behandling ved klassisk Hodgkins lymfom
For barn og ungdom er det viktig at den kumulative dose av hvert enkelt cytostatikum er minst mulig, og at strålefeltene og -dosene reduseres i størst mulig grad. DAL/GPOH gruppen har gjennomført suksessive protokoller der behandlingen er redusert gradvis men tilpasset sykdomsutbredelsen. Kurasjonsratene er opprettholdt. Den avsluttede GPOH-HD 95 protokollen (Dorffel et al., 2013) er nylig publisert med lang oppfølgingtid og viser hva som kan oppnås ved disse protokollene.
I Norge er barn og unge med Hodgkins lymfom siden 2008 behandlet i en europeisk protokoll (EuroNet-PHL-C1). Studien etablerte 2 initiale OEPA kurer (vinkristin, etoposid, prednisolon og doksorubicin) som behandling til alle. I TG 2 og 3 (mer utbredt sykdom) viste en randomisert undersøkelse at videre 2–4 kurer COPDAC-28 (cyclofosfamid, vinkristin, prednisolon og dakarbacin) anses som standard (Mauz-Korholz et al., 2010) (Evidensgrad A). PET-CT ble innført som evaluering før og etter to OEPA kurer som instrument for å avgjøre hvilke pasienter som skal ha supplerende stråleterapi. Ikke publiserte data viser at pasienter med negativ PET-CT etter 2 kurer OEPA (interim PET-CT) som ikke får strålebehandling stort seg har samme helbredelsesrater som pasienter med positiv interim-PET-CT og som gis supplerende strålebehandling (Gallamini et al., 2007) (Evidensgrad C).
Studien med endringer (Pasienter med stadium IA/B eller IIA med SR>30 og/eller største tumorvolum over 200 ml flyttes til TL 2 og COPDAC-28 for alle i TL 2 og 3) danner standardarmene i den nye Euronet-PHL-C2 studien.
Euronet-PHL-C2 studien startet januar 2016. Studien har som mål å redusere bruken av strålebehandling ytterligere ved bruk av mer intensiv kjemoterapi. Det stratifiseres i terapinivåer TL-1, 2 og 3 (se ovenfor). I TL-1 gis OEPA kurer og strålebehandling dersom interim PET-CT er positiv. Om interim PET-CT er negativ, gis en tredje kur som COPDAC-28. I TL-2 og –3 randomiseres til en standard arm der videre kjemoterapi er 2–4 COPDAC-28 eller en eksperimentell arm bestående av 2–4 kurer med tillegg av doxorubicin og etoposid (DECOPDAC-21). For pasienter som er interim PET-CT negative gis ingen strålebehandling. For pasienter som er interim PET-CT positive i standard armen gis strålebehandling etter de samme prinsipper som før. For pasienter med positiv interim PET-CT i den eksperimentelle armen gis kun strålebehandling mot områder med PET opptak etter endt kjemoterapi.
Opptak klassifisert som Deauville score 1–3 regnes som negativt både ved interim-PET–CT og PET-CT etter endt behandling.
Behandling ved residiv
Helbredelse er fortsatt mulig ved første residiv, og i noen tilfeller også ved senere residiver. Prognosen er bedre desto lengre tid etter primærbehandlingen residivet oppstår. Euronet-PHL-C1 studien har en egen residivbehandling som vil egne seg for mange pasienter etter tidligere behandling iht GPOH-HD-95 eller Euronet- PHL-C1/2. Behandlingen innebærer konvensjonelle kurer konsolidert med strålebehandling for pasienter med sene residiver etter den minst omfattende primærbehandlingen, helst også uten strålebehandling primært. For de aller fleste andre vil konvensjonelle kurer etterfulgt av HMAS og eventuelt også strålebehandling være aktuelt ved første residiv (Schellong et al., 2005) (Evidensgrad C). HMAS vil også være aktuelt hos pasienter ved 2. eller senere residiv som ikke har fått HMAS tidligere, og som fortsatt har kjemosensitiv sykdom. Brentuximab vedotin er aktuelt som del av induksjonsbehandlingen og som vedlikeholdsbehandling for en del pasienter med risikofaktorer ved residiv (Moskowitz et al., 2018).
Ved residiv etter HMAS vil behandlingsmålet være å oppnå en ny remisjon for mulig å kunne gjennomgå en allogen stamcelletransplantasjon med familiegiver eller ubeslektet giver. Flere konvensjonelle kurer kan velges, i nyere tid også brentuximab vedotin eller immunterapi med check point inhbiotrer (nivolumab og pembrolizumab (Ansell et al., 2015). Dersom kurativt siktemål ikke lenger er mulig, vil stoffer som gemcitabin, vinorelbin, steroider og trosfosfamid være aktuelle med den hensikt å gi responser, palliasjon og evt. Livsforlengelse, evnt kurer som CEKP eller BOP. Lokal strålebehandling kan være en god palliativ behandling ved kjemoresistent sykdom.
Anbefalinger:
- Barn og ungdom med klassisk Hodgkins lymfom bør tilbys inklusjon i EuroNet-PHL-C2 protokollen, alternativt følge EuroNet-PHL-C1 som terapianbefaling.
- Høydosebehandling med autolog stamcellestøtte kan være aktuell behandling ved residiver.
- Residivbehandlingen har endret seg mye de siste år etter at brentuximab vedotin, nivolumab og pembrolizumab er blitt godkjent for bruk.
- I utvalgte tilfeller av residiver etter høydosebehandling og som på ny har vist god respons på kjemoterapi, kan allogen stamcelletransplantasjon være et alternativ.
Kontrollopplegg for Hodgkins lymfom
Euronet-PHL-C1 studien har spesifikke planer for oppfølging, som i hovedsak gjenspeiler hva en kan anse som rutine for disse pasientene. Kontroller foretas hver 3 mnd i starten, deretter hver 4 mnd, etter hvert hver 6–12 mnd. Klinisk undersøkelse og blodprøver gjøres ved alle kontroller. Billedundersøkelser gjøres ved fastlagte kontrolltidspunkt. Funksjonsundersøkelser av thyreoidea, gonader, hjerte eller lunger samt regelmessig mammografi gjøres etter vurdering av den behandling pasienten har fått, spesielt mht strålebehandling mot de respektive organer. Se Nasjonalt handlingsprogram med retningslinjer for diagnostikk, behandling og oppfølging av maligne lymfomer (Helsedirektoratet, 2019).
Seneffekter, spesielt etter strålebehandling, kan komme sent og pasienter som har fått store strålefelt mot mediastinum bør kontrolleres livslangt. Kontrollene kan etter 5–10. år overføres til lokalsykehus etter retningslinjer gitt fra spesialavdeling, og etter hvert til fastlege. Det er viktig med kompetanse og interesse for denne pasientgruppen ift å påvise residiv tidlig og å kjenne til langtidsbivirkningene etter behandling hos denne relativt unge pasientgruppen (Helsedirektoratet, 2019).
Histiocytoser
Langerhanscellehistiocytose (LCH)
Histiocytoser er en gruppe sykdommer som skyldes unormal proliferasjon av celler tilhørende monocytt-/makrofagsystemet. Ved langerhanscellehistiocytose (LCH) er det langerhanscellene som er den patologiske cellepopulasjonen. Langerhansceller er benmargsderiverte celler som naturlig finnes i hud og lymfeknuter. De prolifererende cellene er vist å være av klonal opprinnelse, men LCH regnes ikke som en kreftsykdom. Den har dog i ICD-10 fått et C-nummer. Nyere molekylær-genetisk forskning fra de siste årene viser i tillegg at en betydelig andel av de patologiske langerhanscellene har mutasjoner i BRAF eller MAP2K genene. Dette har betydning for mulig behandling.
Inndeling
LCH deles inn etter sykdommens utbredelse i enkelt- eller multisystemtype på følgende måte:
- Enkeltsystem-LCH
- Enkeltsystem – én lokalisasjon (single system – single site)
- Enkeltsystem – flere lokalisasjoner (single system – multiple site)
- Enkeltsystem – spesiell lokalisasjon (single system – special site)
- Multisystem-LCH
- Multisystem uten affeksjon av risikoorganer
- Multisystem med affeksjon av risikoorganer (lever, milt, benmarg)
Epidemiologi
Sykdommen er svært sjelden med en anslått insidens på 8–9 tilfeller pr. million barn pr. år, tilsvarende for voksne (Abla, Egeler, & Weitzman, 2010). Det er antatt at dette er et minimumstall, siden enkeltstående lesjoner med spontan tilbakegang ikke alltid blir diagnostisert eller rapportert.
Organmanifestasjoner
- Hud – utslett som ligner seborroisk eksem er vanlig, særlig hos de minste barna. Utslettet er ofte lokalisert i hodebunnen, bak ørene, i øregangene, i hudfoldene i axiller og lysker, nederst på ryggen, i flankene og nederst på abdomen.
- Skjelett – smertefulle lesjoner i skjelettet, ofte i kalvariet eller de lange rørknoklene.
- Lymfeknuter – infiltrasjon av en eller flere lymfeknuter med patologiske langerhansceller. Forstørrede lymfeknuter kan enten opptre som isolert fenomen eller som ledd i mer generalisert sykdom.
- Lever, milt – ofte forstørret som ledd i multisystemsykdom.
- Benmarg – infiltrasjon av benmargen med patologiske langerhansceller. Benmargsaffeksjon som risikokriterium er definert som nedsatt produksjon av hematopoietiske celler (anemi, leukopeni, trombocytopeni).
- Lunger – alene eller som ledd i multisystemsykdom. Lungeaffeksjon er særlig vanlig hos voksne røykere
- Tarm – tarmaffeksjon er sjelden, men sees iblant.
- CNS
- Diabetes insipidus (DI) er svært vanlig ved LCH. DI kan være første manifestasjon, ledd i multisystemsykdom eller sees ved enkeltsystem skjelettsykdom. DI er særlig hyppig som komplikasjon ved skjelett-LCH lokalisert i skallebasis eller ansiktsskjelett.
- Nevrodegenerativ LCH – dette er en tilstand som rammer noen LCH-pasienter. Det er uklart hva det egentlig representerer, ikke nødvendigvis aktiv LCH
Symptomer
Symptomene varierer med sykdommens lokalisasjon og utbredelse.
- Enkeltsystem-LCH:
Det er først og fremst lokalisasjonen som avgjør symptomene: utslett, smertefull kul/hevelse, hovne lymfeknuter etc., se foregående avsnitt - Multisystem-LCH
Her er mer enn ett organsystem affisert, med eller uten allmennsymptomer, med eller uten affeksjon av risikoorganer
Risikoorganer:- Lever
- Milt
- Benmarg (definert som cytopeni i minst 2 linjer)
Tidligere var også lunger definert som risikoorgan, men dette er i den nyeste protokollen tatt ut som risikoorgan.
Karakteristisk for sykdommen er tendensen til spontane remisjoner og eksaserbasjoner.
Diagnose
Radiologisk utredning ved diagnose er røntgen totalskjelett, røntgen thorax og ultralyd abdomen. Ytterligere undersøkelser som CT av lungene og CNS utredning kun på indikasjon.
Diagnosen stilles ved biopsi av en lesjon. De patologiske cellene ved LCH skiller seg fra normale langerhansceller ved at de på overflaten har markøren CD1a, og at de inneholder Birbeck-granula som er synlige ved elektronmikroskopi. For en sikker diagnose kreves påvisning av CD1a og/eller CD207 (Langerin) positive celler. Elektronmikroskopisk påvisning av Birbeck-granula er ikke obligatorisk.
Ved DI som første manifestasjon av LCH vil vi ofte finne en fortykket hypofysestilk ved MR-undersøkelse. Før evt. spesifikk LCH-behandling kreves biopsiverifikasjon.
Behandling
Behandlingen har tradisjonelt vært kontroversiell pga. sykdommens naturlige svært svingende forløp. Det har vært to prinsipielt forskjellige tilnærminger til behandlingen, dels et behandlingsprinsipp som innebærer så lite behandling som mulig pga. den store tendensen til spontane remisjoner, dels et prinsipp om tidlig og intensiv behandling med kjemoterapi for at sykdommen skal falle til ro raskt, for å redusere permanente sekveler og redusere risikoen for reaktivering av sykdommen. Gjennom Histiocyte Society (stiftet i 1985, en internasjonal organisasjon for forskning og behandling av histiocytoser) har man etter hvert lyktes i å komme frem til felles behandlingsretningslinjer basert på resultater fra tidligere studier. Siden 2015 har vi behandlet pasienter i henhold til LCH IV protokollen fra Histiocyte Society.
- Enkeltsystemsykdom
Behovet for aktiv behandling varierer avhengig av hvilket organ som er affisert, og hvilken eller hvilke lokalisasjoner- Hud: Lokal steroidbehandling, evt. peroral etoposid (bare aktuelt til voksne). Hud-LCH kan være ledd i multisystemsykdom, slik at pasienten må utredes med henblikk på andre lokalisasjoner. Evt. kan det skje videreutvikling til eller reaktivering i form av multisystem-LCH.
- Skjelett: Avhengig av lokalisasjon og om det er en enkelt lesjon eller flere. En enkelt lesjon vil ofte gå tilbake spontant eller etter diagnostisk biopsi, evt. injeksjon av lokale steroider. Visse lokalisasjoner krever aktiv behandling for å avverge senskader (f. eks. lesjon i virvelcorpus med risiko for skade av medulla spinalis, lesjoner i skallebasis eller ansiktsskjelett med økt risiko for CNS-affeksjon). Dette er spesifisert i protokollen. Indometacin og bisfosfonater har også vært prøvd ved skjelett-LCH, og indometacin er et behandlingsalternativ i en randomisert arm i LCH-IV. De enkelte lesjoner følges med røntgen i henhold til den aktuelle protokoll.
- Isolert diabetes insipidus: DI som ledd i LCH er som regel irreversibel. DI behandles med desmopressin. Man må ikke la seg friste til å gi spesifikk LCH-behandling uten biopsiverifikasjon. Siden man som regel vegrer seg for å ta biopsi av en lesjon i hypofysestilken, må man lete etter andre lokalisasjoner for biopsitagning, undersøke spinalvæsken med henblikk på tumormarkører (diff.diagnose: germinalcellesvulst) og CD1a pos. celler. Ved verifisert LCH og en tydelig hypofysestilktumor er det indikasjon for systemisk behandling med vinblastin/steroider etter LCH-IV-protokollen.
- Multisystemsykdom (samme behandling for MS-LCH med eller uten riskorgan affeksjon):
Behandling etter LCH-IV protokollen stratum 1. Behandlingen består av en eller to induksjonsblokker med vinblastin x 6 og steroider per os, etterfulgt av en vedlikeholdsbehandling som inneholder pulser med vinblastin/steroider, randomisert med eller uten 6-merkaptopurin og varighet 12 eller 24 måneder (dvs. 4 randomiseringsarmer). - Multisystemsykdom med forverring eller reaktivering av ikke-risiko organer:
Behandling etter LCH-IV protokollen stratum 2. Behandlingen består av behandling med steroider, cytarabin og vinkristin i 24 uker, deretter randomisering til tablettbehandling med indometacin eller purinethol/metotreksat til totalt 2 års behandling. - Multisystemsykdom med forverring eller reaktivering av risikoorganer:
Det er dette som i størst grad tilsvarer den gamle betegnelsen «Letterer-Siwe disease». Dette er den mest alvorlige formen for LCH, og det er i denne gruppen vi finner pasienter med livstruende sykdom og en relativt høy mortalitet. I LCH-II-protokollen (1996–2000) var mortaliteten 75 % hos pasienter som ikke responderte på behandlingen i løpet av 6–12 uker. Disse pasientene behandles nå i henhold til LCH IV protokollens stratum 3. Behandlingen består av høye doser cytarabin og cladribin, supplert etterhvert med purinethol, metotreksat, vinblastin og steroider. Dersom det ikke er respons på denne behandlingen kan allogen benmargstransplantasjon være et behandlingsalternativ.
BRAF/MEK hemmere
Det er økende evidens for god behandlingsrespons med BRAF hemmere (og også etterhvert MAP2K hemmere) der man finner tilgrunnliggende BRAF/MAP2K mutasjoner i langerhanscellene. Derimot har man sett at pasientene har tendens til reaktivering når man avslutter behandlingen. Disse medikamentene er pr i dag ikke innlemmet i LCH IV protokollen, og det er foreløpig ikke nok kunnskap til å vite hvilke pasienter som bør tilbys behandling, og hvor lenge behandlingen bør vare. I dag blir behandling med BRAF/MAP2K hemmere først og fremst anvendt der man ikke oppnår tilfredsstillende behandling med konvensjonell behandling.
Radiologisk oppfølging
Ved CNS risikolesjoner hver 3. måned første år, hver 6 måned 2.–5. År. Ved skjelettlesjoner røntgen hver 3. måned til tilbakegang. Ytterligere oppfølging kun på indikasjon
Prognose
For enkeltsystem-LCH og multisystem-LCH uten affeksjon av risikoorganer er prognosen vedrørende overlevelse svært god (100 %). Det er dog en stor risiko for en eller flere reaktiveringer av sykdommen og for permanente sekveler, først og fremst ortopediske, diabetes insipidus og evt. panhypopituitarisme. Det er også de senere årene blitt klart at enkelte pasienter kan få en nevrodegenerativ tilstand forårsaket av LCH. Denne tilstanden er lite påvirkelig av terapi. I LCH IV protokollen anbefales nå at denne gruppen pasienter med kliniske symptomer på nevrodegenerasjon behandles med enten cytarabin eller immunglobuliner.
Hemofagocytisk lymfohistiocytose (HLH)
Hemofagocytisk lymfohistiocytose (HLH) betegner en hyperinflammatorisk tilstand karakterisert ved langvarig høy feber, cytopenier, hepato-/splenomegali og hemofagocytose i aktiverte, benigne makrofager. HLH kan være primær/genetisk eller sekundær til andre sykdommer, først og fremst infeksjoner, maligne tilstander eller revmatiske lidelser. HLH forekommer både hos barn og voksne. Primær/genetisk HLH opptrer ofte svært tidlig, gjerne i første leveår, mens sekundær HLH vanligvis opptrer senere (Janka, 2007).
Felles for både primær og sekundær HLH er at fagocyterende celler (cytotoxiske T‑lymfocytter (CTL) og NK-celler) som aktiveres etter stimulering av et (infeksiøst) agens, produserer store mengder inflammatoriske cytokiner, noe som normalt vil medføre at det aktuelle patogene agens blir fjernet. Ved HLH har NK- cellene og CTL defekt fagocyterende evne, og cytokinnivået vil derfor holde seg vedvarende høyt og vedlikeholde den inflammatoriske tilstanden.
Diagnosen HLH er ofte vanskelig. Tilstanden omtales her fordi den kan være en vanskelig differensialdiagnose til leukemi hos barn, og fordi den behandles av spesialister i barnehematologi/-onkologi med cytostatika og hematopoietisk stamcelletransplantasjon.
Primær/familiær HLH (FHLH eller FHL) Tilstanden er autosomalt recessivt arvelig med en hyppighet på 1,2:1.000.000 barn pr. år eller ca. 1: 50.000 levende fødte barn i Sverige (Henter, Elinder, Soder, & Ost, 1991). Det er til nå identifisert fire ulike gendefekter som kan gi HLH, mens det hos 50–70 % av pasientene (avhengig av etnisk bakgrunn) ikke kan påvises en kjent gendefekt. Diagnosen er vanskelig, og spesielt tidligere ble diagnosen i en del tilfeller stilt post mortem. Symptomene opptrer ofte i svært ung alder, gjerne tidlig i første leveår, men er også beskrevet høyere opp i barnealder. Selv i arvelige tilfeller er tilstanden ofte utløst av en infeksjon, slik at distinksjonen mellom primær/arvelig og sekundær HLH kan være vanskelig. HLH kan også opptre som ledd i arvelige immunsvikttilstander: Chédiak-Higashis syndrom (CHS), Griscellis syndrom (GS) og X-bundet lymfoproliferativt syndrom (XLP).
Sekundær/reaktiv HLH Tilstanden opptrer sekundært til infeksjoner («virus associated hemophagocytic syndrome» – VAHS (særlig EBV), eller «infection associated hemophagocytic syndrome» – IAHS), maligne sykdommer eller revmatiske lidelser (representerer en litt annen entitet, «macrophage activation syndrome»– MAS). De fleste tilfellene opptrer hos litt større barn eller voksne. Epstein- Barr-virus er antagelig den vanligste utløsende årsaken ved IAHS. Visceral leishmaniasis er en vanskelig og viktig differensialdiagnose, ikke minst fordi infeksjonen er i ferd med å bre seg i Europa (Ready, 2010). Leishmaniainfeksjon kan også være en årsak til sekundær HLH. Hemofagocytose kan også sees som et mer uspesifikt reaktivt fenomen, f. eks. hos intensivpasienter med multiorgansvikt (Strauss et al., 2004).
Diagnostiske kriterier (enten 1. eller 2. må være oppfylt) (Henter et al., 2007):
- Molekylær diagnose forenlig med HLH
- Diagnostiske kriterier for HLH til stede (minst 5 av 8):
- Opprinnelige kriterier:
- Feber
- Splenomegali
- Cytopeni i minst 2 cellelinjer i perifert blod
(Hb < 9 g/dL, tr.c. < 100 x 109/L, nøytrofile < 1,0 x 109/L) - Hypertriglyceridemi og/eller hypofibrinogenemi
(fastende triglycerider ≥ 3,0 mmol/L, fibrinogen ≤ 1,5 g/L) - Hemofagocytose i benmarg, milt eller lymfeknute
(ofte ikke til stede tidlig i sykdomsforløpet). Ingen tegn til malignitet
- Nye kriterier:
- Lav eller opphevet NK-celleaktivitet
- Ferritin ≥ 500 μg/L
- Løselig CD25 (IL-2-receptor) ≥ 2400 U/ml
- Opprinnelige kriterier:
Hemofagocytose som har gitt sykdommen dens navn, er ofte ikke til stede tidlig i forløpet, og det kan også sees ved andre tilstander. Det er derfor viktig å ha en høy grad av mistenksomhet overfor HLH hos alvorlig syke, septiske sped- og småbarn som har cytopenier, hepato-/splenomegali og manglende effekt av antibiotikabehandling. Mange har CNS-affeksjon med forhøyet celletall i spinalvæsken. Det er avgjørende at de riktige prøvene blir tatt, først og fremst ferritin, triglycerider og koagulasjonsstatus inkludert fibrinogen. Undersøkelse av interferon og interleukiner kan være til hjelp for å stille diagnosen, (IL-2 reseptor analyseres bl.a på Hormonlaboratoriet på Aker (OUS) (0.5 ml frossent serum)). Det må også gjøres benmargsundersøkelse for å utelukke leukemi og eventuelt påvise hemofagocytose.
Behandling
Eventuell utløsende infeksjon må behandles. Ved EBV infeksjon, kan Rituximab være av nytte. EBV-indusert HLH kan bli svært alvorlig og få dødelig utfall.
Det er utarbeidet en protokoll for behandling av HLH gjennom Histiocyte Society (Henter et al., 2007). Den første protokollen kaltes HLH-94-protokollen, den er senere erstattet med HLH-2004-protokollen der det bare er gjort relativt små endringer.
Behandlingen består av steroider (dexametason) kombinert med etoposid og cyklosporin A i induksjonsfasen over 8 uker. Ved CNS-affeksjon som vedvarer etter 2 ukers behandling, gis intratekal behandling med metotreksat og prednisolon. Ved primær HLH går man etter 8 uker rett over i en vedlikeholdsfase med de samme medikamentene, frem til det kan utføres hematopoietisk stamcelletransplantasjon (SCT) som er eneste kurative behandling. Dersom man ikke oppnår remisjon blir 2. linje behandling forsøkt, f.eks tillegg av antithymocytt-globulin, og individuell tilpasning av behandlingen må gjøres. Hvis man mistenker at det er en sekundær HLH (mildere forløp, høyere alder, ingen familieanamnese eller kjent gendefekt), vil man seponere behandlingen etter 8 uker dersom det er god behandlingsrespons, og følge nøye opp om det kommer et residiv eller ikke. Ved dårlig behandlingsrespons eller tilbakefall må man behandle som primær HLH med SCT, men mange vil vise seg å være helbredet etter de første 8 ukene. Ved mindre alvorlige tilfeller av sekundær HLH kan det være tilstrekkelig med god støttebehandling og immunmodulerende behandling, men terskelen for å behandle etter HLH-2004-protokollen må være lav, siden også den sekundære formen kan ha et raskt progredierende forløp med fatal utgang. HLH-protokollen ble lukket ved årsskiftet 2011–2012, men det anbefales at den brukes fremdeles med en liten modifikasjon i form av utsatt start av cyklosporin til dag 14.
Per februar 2020 pågår det en metodevurdering av Emapalumab (anti-interferon-gamma) til bruk ved refraktær primær HLH. Dette medikamentet ble FDA-godkjent november 2018, og det er en pågående pediatrisk investigational plan («PIP») i Europa. Ved evt behov for medikamentet i en refraktær situasjon før markedsføringstillatelse, kan man henvende seg til produsent for å forhøre seg om muligheten for Compassionate Use, etter retningslinjer i Helseforetaket.
Prognose
Før behandlingen etter HLH-protokollene ble etablert, var den primære formen for HLH alltid fatal, men med behandling som inkluderer SCT, overlever over 50 % av pasientene. Også sekundær HLH kan ha et alvorlig forløp med fatal utgang.
Svulster i sentralnervesystemet
Sist faglig oppdatert: 26.05.2020
Lavgradig gliom
Lavgradige gliomer (LGG) er fellesnavn på svulster utgående fra gliaceller med malignitetsgrad 1 og 2 etter WHO-klassifiseringen (Louis et al., 2007). Hos barn er nesten alle disse astrocytomer. LGG skiller seg fra høymaligne svulster ved at tumorveksten hos de fleste er langsom, og hos noen kan endog veksten stoppe spontant uten behandling. Forløpet kan være fluktuerende med periodevis økning og regresjon av tumorstørrelse, hos andre ses et mer aggressivt forløp og endog metastasering til hjernehinner. Som følge av LGGs natur har pasientene en meget høy overlevelse (OS, Overall Survival), mens progresjonsfri overlevelse (PFS) er mye lavere, dvs at progresjon er vanlig, men lar seg oftest vellykket behandle. Barn under ett år har tendens til mer negativt forløp enn eldre barn. Forskjellige histopatologiske undergrupper av LGG har også forskjellig prognose. Svulstenes lavgradige natur er en meget viktig faktor ved vurdering av behandlingsvalg.
Det er nylig publisert europeiske retningslinjer for diagnose og behandling av LGG hos barn og ungdom (A. K. Gnekow et al., 2019).
Epidemiologi
LGG er de vanligste CNS-svulster hos barn og utgjør ca. 40 % av disse. Pilocytisk astrocytom (PA) er den hyppigst forekommende svulst av alle typer registrert i barnekreftregisteret, ca. 10 % av alle pasienter totalt registrert (Stiller, 2007).
LGG er sterkt assosiert med nevrofibromatose 1 (NF-1), og NF-1-pasienter har særlig overhyppighet av pilocytiske astrocytomer i synsveiene (A.K. Gnekow, Packer, & Kortmann, 2004; Stokland et al., 2010). Ofte er disse multifokale med involvering av hjernestammen og andre strukturer.
Histopatologiske typer og lokalisering
Den helt dominerende typen LGG hos barn er pilocytiske astrocytomer (PA) (WHO grad 1) som utgjør over 80 % av gruppen(Stokland et al., 2010). Diffuse astrocytomer grad 2 (DA) er vanligst hos voksne, men utgjør knapt 10 % hos barn, noe hyppigere hos ungdommer. En variant av PA, pilomyxoid astrocytom (PMA) (WHO grad 2) har et mer aggressivt forløp.
Oligodendrogliomer er svært sjeldne hos barn. Diffuse ponsgliomer inkluderes ikke i LGG selv om enkelte av disse histololgisk er DA (WHO grad 2).
Blandingssvulster av nevronale og gliale celler er sjeldne. De er nesten alltid histologisk lavgradige og omtales oftest sammen med LGG. Den vanligste typen er gangliogliom.
LGG kan oppstå i alle deler av CNS, mest hyppig i cerebellum, synsveiene (n. opticus, chiasma, sentrale synsbaner) og hemisfærene (Stokland et al., 2010). Lokalisasjon varierer med alder. Små barn har overvekt av synsveigliomer mens større barn tenderer mot hemisfærebeliggenhet. Pilocytiske astrocytomer er den dominerende typen i alle aldersgrupper, mens fibrillære er noe hyppigere hos ungdommer enn hos små barn. I synsveier og cerebellum finnes nesten utelukkende PA, mens DA utgjør ca. 10–15 % i andre lokalisasjoner.
Molekylærbiologi
De senere år har en funnet svært viktige forandringer molekylærbiologisk hos barn med LGG (Packer et al., 2017). Det gjelder særlig i signalveien MAPK (RAS/RAF/MEK/ERK) som er viktig for cellenes proliferasjon. Spesifikke endringer i denne signalveien kan gi overaktivitet og dermed økt onkogen stimulering. Pilocytisk astrocytoms biologi regnes i dag å representere en «singel pathway disease». Fusjon av BRAF-genet med annet gen (BRAF-KIAA1549) er vanligst ved PA, mens punktmutasjon av BRAF (BRAF V600E) er sjeldnere og ses ved forskjellige LGG. Det foreligger allerede i dag medikamenter som har LGG med slike mutasjoner som mål (MEK-inhibitorer og BRAF-inhibitorer). Se senere.
Diagnose og utredning
LGG gir symptomer enten pga lokal påvirkning eller pga økt intrakranielt trykk, og bildet vil variere sterkt avhengig av lokalisasjon. Synsfunksjonen er ofte påvirket ved supratentorielle midtlinjesvulster og n. opticus-gliomer. Balanseforstyrrelser ses ved cerebellare svulster, epilepsi ved hemisfærelokalisasjon etc. Økt intrakranielt trykk gir hodepine mest uttalt om morgenen, morgenkvalme og brekninger og noen ganger dobbeltsyn (abducensparese). Hvis symptomene har vart lenge er det tegn på lavgradig svulst, men kort varighet er ikke uvanlig.
MR cerebrum viser tumor. På tross av histopatologisk lavgradighet, kan LGG være disseminert eller multifokal, og derfor skal hele CNS visualiseres. Anbefalte MR-teknikker er som ved andre hjernesvulster hos barn.
Histologisk diagnose er oftest nødvendig, og når radikal reseksjon ikke er mulig, gjøres biopsi. Unntak er synsveigliomer med klassiske MR-funn, særlig hos nevrofibromatose-pasienter, der man godtar radiologisk diagnose uten biopsi, fordi disse svulstene erfaringsmessig alltid er PA. Ved tectum-lokalisasjon vil en også være tilbakeholdende med biopsi.
Molekylærbiologisk utredning er ønskelig ved de fleste LGG både for diagnose og valg av behandling.
Oftalmologisk utredning er svært viktig ved supratentorielle midtlinjegliomer, spesielt i synsveiene. Endokrinologisk kartlegging er vesentlig avhengig av lokalisasjon.
Behandling
Behandling og prognose vil variere med lokalisasjon. Laterale svulster vil ligge bedre til rette for radikal kirurgi, og dermed ha bedre prognose. Midtlinjesvulster vil ikke kunne fjernes radikalt og trenger derfor i mange tilfeller ytterligere behandling, ut fra klare retningslinjer (Perilongo, 2005; Reddy & Packer, 1999).
Kirurgi er primærbehandlingen ved LGG (B. J. Due-Tønnessen et al., 2002). Reseksjonsgrad er den viktigste prognostiske faktor, og radikal reseksjon gir best prognose. Ofte vil radikal kirurgi ikke være mulig pga faren for skade, og da synes ikke partiell reseksjon å gi resultatmessige fordeler vedrørende overlevelse sammenlignet med biopsi (Stokland et al., 2010). Men i en del tilfeller vil partiell reseksjon være hensiktsmessig for å bedre pasientens kliniske tilstand, f.eks. ved hydrocephalus. Tilnærmingen må ses i forhold til mulige skadelige effekter av en partiell reseksjon.
Norge har deltatt i den store internasjonale behandlingsprotokollen SIOP-LGG 2004, som ble avsluttet i 2015. En ny studie (LOGGIC) er nå (2019) på det nærmeste ferdigutformet av SIOP-Europe med randomisering i tre behandlingsarmer. Kombinasjonen Vinkristin/Karboplatin er standardarmen, Vinblastin monoterapi som er mye brukt i dag er andre arm, og den mest eksperimentelle armen vil være MEK-inhibitor (Trametinib). Trametinib gis peroralt. LOGGIC åpner i Norge i 2020. NF1-pasienter inkluderes sannsynligvis ikke i denne fordi denne gruppen skal rekrutteres til en annen studie. Ca. 1/3 av alle LGG-pasienter forventes å trenge ikke-kirurgisk behandling. Det er viktig at klare kriterier oppfylles før slik behandling startes. Dette innebærer at ved diagnose / rett etter primærkirurgi, gis medikamentell behandling bare hvis pasienten har alvorlige nevrologiske symptomer som kan forverres, eller truende synstap. Alle andre pasienter blir observert klinisk og billedmessig. Disse pasientene, som utgjør et flertall, er observasjonsgruppen i studien. Dersom tumor i observasjonstiden vokser signifikant ved MR-undersøkelse, eller det er økende symptomer, vurderes da på ny evt kirurgi. Dersom dette ikke er hensiktsmessig, inkluderes også disse pasientene.
Da også de behandlingstrengende pasientene har god prognose vedrørende overlevelse, innfører LOGGIC nye primære endepunkter, særlig gjelder dette synsfunksjonen. Effektiv behandling som bevarer synet vil være sterkt ønsket. Synsundersøkelse med analyse av funksjonsendring vil derfor være en hovedfaktor i studien.
LGG-pasienter med nevrofibromatose (NF-1) planlegges inkludert i et felles studium med USA. Det ligner LOGGIC, men har bare to armer: vinkristin/karboplatin vs. MEK-inhibitor. Dersom studien ikke lar seg gjennomføre, vil gruppen bli inkludert i LOGGIC.
Strålebehandling kan ha god effekt ved LGG og var tidligere mye brukt. Pga mulige kognitive seineffekter og sekundærkreft, er radioterapi nå ikke en del av primærbehandlingen. Imidlertid kan det være aktuelt når medikamentell behandling ikke er effektiv nok, spesielt ved små svulster og hos eldre barn og ungdom. Strålekniv kan brukes i utvalgte tilfeller. Hos NF-1-pasienter vil en være enda mer tilbakeholdende med strålebehandling pga økt følsomhet for alvorlige strålebivirkninger
En oppnår sjelden komplett remisjon av medikamentell behandling, men varig stabil tumorstørrelse eller noe reduksjon vurderes som tilfredsstillende behandlingseffekt.
Anbefalinger:
- Kirurgi anbefales når radikal tumorreseksjon er mulig, og denne behandlingsmodus skal alltid vurderes først.
- Når radikal kirurgi ikke er mulig, skal biopsi gjøres i de aller fleste tilfellene. Partiell reseksjon synes ikke å gi bedre overlevelsesresultater enn biopsi og er derfor omdiskutert. Dette må tas stilling til i hvert enkelt tilfelle.
- Kirurgi vurderes på ny ved residiv eller progresjon.
- Når klare kriterier er oppfylt (se ovenfor), anbefales å gi nonkirurgisk behandling i henhold til LOGGIC protokollen.
- Pasienter med nevrofibromatose behandles etter egen protokoll.
Ny utprøvende behandling
MEK-inhibitor hemmer MAPK-signalveien «downstream» for BRAF og har vist seg å gi god respons ved LGG i ferske studier. Dette har ført til at trametinib nå skal brukes «up-front» i LOGGIC-studien. Langtidseffekten er foreløpig ikke kjent, men ved behandlingsrefraktær LGG med dårlig prognose bruker mange nå MEK-inhibitor, peroralt en gang daglig. En forventer etter hvert mer kunnskap om virkninger på lengre sikt.
BRAF-inhibitor gir god respons på svulster som har BRAF-punktmutasjon, og er i dag førstevalg når slik mutasjon er påvist. Imidlertid gir den paradoksal effekt i form av økt tumoraggressivitet ved svulster med BRAF-fusjon, og må derfor aldri gis uten bekreftet BRAF V600E punktmutasjon.
Bevacizumab alene eller kombinert med andre midler brukes ved resistent LGG i dag. Den gir god respons hos tidligere sterkt behandlede pasienter, men nesten alle får progresjon etter avsluttet behandling(Hwang et al., 2013). Positiv påvirkning av visus har vært rapportert, men flere studier har ikke kunnet verifisere dette.
Behandling av tilbakefall eller progresjon etter primærbehandling
LGG har et helt annet forløpsmønster enn aggressive kreftformer. Ofte starter tumor ny vekst flere år etter at den har blitt stabilisert av behandling. En kan da gi samme behandling som initialt, eller andrelinjebehandling. I Norge er det for tiden vanligvis vinblastin monoterapi. MEK-inhibitor kan vurderes som anført ovenfor.
Anbefalinger:
- Dersom primærbehandlingen hadde god effekt, kan en gjenta denne.
- Ved tidlig progresjon anbefaler vi annenlinjebehandling. Mest brukt nå er ukentlig vinblastin i ett år (Bouffet et al., 2012).
Organisering av behandlingen
Behandlingen av LGG er regionalisert og ledes av det regionale barnekreftsenteret. Dette gjelder både kirurgi, kjemoterapi og strålebehandling. Deler av kjemoterapien kan gis mer lokalt.
Prognose
Mortaliteten er lav med 5 års overlevelse rundt 95 %, mens progresjonsfri overlevelse (PFS) er atskillig lavere. Komplett reseksjon er den viktigste uavhengige positive prognostiske faktoren, mens alder under ett år ved diagnose og de viktigste grad 2-histopatologiske typene (DA og PMA) er negative faktorer. Ved svulster i synsveiene har NF1-pasienter bedre prognose enn andre (Stokland et al., 2010). Ved synsveigliomer får pasientene ofte senskade i form av betydelig og varig synsreduksjon. DA kan ha en tendens til å utvikle seg til malignt astrocytom etter flere år, men i langt mindre grad enn hos voksne. Dette skjer svært sjelden ved PA, men er beskrevet.
Oppfølging og seineffekter Anbefalinger:
- Klinisk kontroll med MR gjøres hver tredje måned de første to år, deretter hver 6. måned, og deretter årlig.
- Progresjon kan oppstå etter flere år, slik at oppfølging i ca. 10 år anbefales.
- Oftalmologisk oppfølging er essensielt hos pasienter med synspåvirkning.
- Nevropsykologisk testing gjennomføres 1, 2 og 5 år etter diagnose ved regionsykehusene i hht nasjonalt tverrfaglig oppfølgingprogram for barn med tumor cerebri, og det gjøres en vurdering av behov for utredning også tidligere i forløpet. Pedagogisk oppfølging ved behov er vesentlig.
- Habilitering og andre former for oppfølging individualiseres.
Høygradige gliomer og Diffuse ponsgliomer
Høygradige gliomer (HGG) er de vanligste CNS-svulster hos voksne, men betydelig sjeldnere hos barn. De aller fleste er astrocytomer, og etter oppdatert WHO- klassifiseringen av CNS svulster fra 2016 skilles det mellom anaplastisk astrocytoma IDH wild type eller IDH mutert (AA) (WHO grad 3), glioblastoma (GB) IDH wild type eller IDH mutert (WHO grad 4) og diffuse midtlinje gliomer, H3K27M-mutert (WHO grad 4) (Gupta & Dwivedi, 2017). Høygradige astrocytomer hos barn har en annen mutasjonsspektrum sammenlignet med voksne svulster. For eksempel viser anaplastiske astrocytomer hos barn veldig sjelden IDH mutasjon som er mer typisk hos voksne. Diffuse ponsgliomer (DIPG- Diffuse Intrinsic Pontine Glioma) er en undergruppe av diffuse midtlinje gliomer og omtales separat da de har egen behandling på grunn av spesifikk biologi og klinisk forløp med svært dårlig prognose.
Diffuse/Fibrillære astrocytomer (WHO grad 2) er lavgradige gliomer, men det kan være en vanskelig avgrensning mot grad 3, og svulstene kan utvikles i malign retning med tiden.
Vi finner HGG i hele CNS, fra 35–50 % supratentorelt, 40–50 % oppstår i hjernestammen inkludert ponsområdet som diffuse ponsgliomer(DIPG), i midtlinjestrukturer inkludert thalamus (13 %), i lillehjernen (5 %) og i spinalkanalen (3 %) (Gianno F, 2018).
Epidemiologi
HGG har en insidens på ca. 0,85 pr 100 000 barn og ungdom fra 0–19 år pr år og utgjør ca. 8–12 % av alle CNS-svulster hos barn (Braunstein, Raleigh, Bindra, Mueller, & Haas-Kogan, 2017).
Strålebehandling mot CNS fører på sikt til en økt risiko for utvikling av HGG.
Patologi
Både AA og GB vokser diffust infiltrativt og tumor kan være vanskelig å avgrense på MR bilder. Begge svulstene har høy mitotisk aktivitet, høy cellularitet, kjerneatypi og cellulær pleomorfisme. Hos GB finner vi i tillegg varierende områder med nekrose, småblødninger, fast og bløtt vev, i tillegg til mikrovaskulær proliferasjon.
Skillet mellom grad 3 (AA) og grad 2 (DA/FA) kan være vanskelig, spesielt i små biopsier, og patologens diagnostisering vil influere betydelig på resultatene i studier for hhv HGG og LGG etter hvordan grensetilfellene blir vurdert.
Molekylærbiologi
De siste årene har det vært en voldsom økning i kunnskapen om molekylærbiologiske forhold ved hjernesvulster og for HGG er det funnet stor heterogenitet. Kartlegging av arvematerialet ved HGG har bl.a. påvist mutasjoner som affiserer histonfunksjonen i svulstcellene (K27M og G34R/V mutasjon i H3.3 og H3.1), og som er et kjennetegn ved HGG hos barn og unge voksne. Videre er det funnet molekylære subgrupper (for.eks. H3.3 K27M, H3.1 K27M, H3.3 G34R/V) som kan si noe om svulstens plassering i CNS, typisk alder når svulsten opptrer og om prognose.
I kortikale svulster finnes ofte mutasjon eller overekspresjon av TP53, i tillegg til metylert MGMT og 5–10 % av svulstene i dette område utrykker BRAF V600E mutasjon.
Det finnes også andre genetiske forandringer som epidermal vekstfaktor-reseptor (EGFR) og platederivert vekstfaktor-reseptor A (PDGFRA) som kan være mutert og/eller ha amplifikasjon/overekspresjon i ulike sub-grupper av HGG (Sturm, Pfister, & Jones, 2017).
Diagnose og utredning
Som ved de fleste andre hjernesvulster avhenger symptomene ikke av histologi, men mest av tumors lokalisasjon som forårsaker lokal påvirkning eller økt intrakranielt trykk. Ofte har symptomene vart i kort tid pga. svulstenes aggressive vekst. Hodepine, kvalme/oppkast, epilepsi og hemiplegi er vanlige symptomer. Andre fokalt fremkalte symptomer forekommer. Rask forverring skjer hvis pasienten ikke blir behandlet.
Ved MR undersøkelse av HGG er tumor uklart begrenset med irregulære marginer. Som oftest ses inhomogen kontrastoppladning. GB viser oftest sentral nekrose omgitt av en hyperintens sone og ødem rundt tumor er vanlig.
Tumor er som oftest fokal, men kan gi spinal spredning, slik at hele CNS skal visualiseres.
Histopatologisk diagnose er helt nødvendig, og materiale fås ved reseksjon eller biopsi.
Behandling
Kirurgi er en svært viktig del av behandlingen, og målet er å fjerne hele svulsten radikalt hvis mulig, eventuelt så mye som mulig uten for stor skade. Da svulsten har et infiltrativt vekstmønster, vil det alltid være gjenværende tumorceller rundt reseksjonen.
Fokal strålebehandling gis til alle barn over 3 år. HGG har ikke mikrospredning til CNS slik at stråling av hele CNS er ikke nødvendig. Senskadene blir dermed mindre.
Kjemoterapi
Allerede i 1976 ble det i USA startet behandling med kjemoterapi bestående av prednison, lomustin og vincristin (pCV), med signifikant bedring av resultatene i forhold til stråling alene. Det samme ble vist i andre studier hvor kjemoterapi i tillegg til strålebehandling gav økt overlevelse i forhold til bare stråling (Sposto et al., 1989). Senere har det i flere studier vært ny histologisk gjennomgan av tumorvev som har vist at mange lavgradige gliomer har vært inkludert i studiene, og at kjemoterapi i tillegg til kirugi og stråling ikke gav bedre overlevelse ved HGG (Fouladi et al., 2003).
Senere utprøving av nye regimer har ikke klart å bedre resultatene for HGG. Temozolomid sammen med og etter strålebehandling har hatt en viss effekt hos voksne, men det er ikke funnet samme effekt hos barn(K. J. Cohen et al., 2011).
Det prøves ut en rekke nye stoffer og prinsipper i behandlingen av HGG hos barn. Lovende resultater har blitt oppnådd med vaksinasjon med dendritiske celler. Ut fra økende molekylær forståelse av HGG hos barn prøves det ut stoffer som påvirker forskjellige molekylære signalveier, i form av monoklonale antistoffer, småmolekylinhibitorer, strålesensitizere, genterapi og immunterapi. Disse kan påvirke celleproliferasjon, angiogenese, invasivitet og celleoverlevelse (Hargrave, 2009). Rebestråling av supratentoriale HGG har vist forlenget levetid sammenliknet med barn som ikke fikk rebestråling (Tsang, Oliveira, et al., 2019).
For øyeblikket er det ingen åpne studieprotokoller for barn med HGG. Internasjonalt er det ikke konsensus om beste behandling. De fleste barn i Norge får kombinasjonsbehandlingen med stråling og Temozolomid eller stråling og to ulike kjemoterapi, Temozolomid og Lomustine. Den siste kombinasjonen har vist seg å være mer effektiv enn Temozolomid alene, spesielt hvis tumorvevet utrykkeraktiv (umetylert) MGMT enzym promotor som medfører begrenset effekt av Temodal (Jakacki et al., 2016).
HGG hos barn < 3 år er veldig sjeldent og de har en bedre prognose sammenliknet med eldre barn, grunnet ulik biologi i svulstvevet (Gielen et al., 2015). Disse barna har nytte av kjemoterapi og anbefales behandling etter «SIOP Infant HGG protocol».
Prognose
Det har vært lite fremgang i resultatene for HGG. 5 års overlevelse ligger mellom 15 og 25 %. Histologi og omfanget av tumorreseksjon er viktige prognostiske faktorer, i tillegg til proliferasjonsindex. AA har bedre prognose enn GB og total tumorreseksjon og lavere proliferasjons index gir bedrer prognosen. I omtrent 40 % av HGG påvises mutasjon i tumor supressor genet, TP53, noe som er forbundet med dårlig prognose. Påvisning av mutasjoner i H3F3A, genet som koder for histonfunksjonen i svulstcellene, gir opphav til ulike molekylærbiologiske subgrupper som kan si noe om svulstens plassering i CNS, typisk alder når svulsten opptrer og om prognose. Svulster med H3.3 K27M mutasjon og H3.1K27M mutasjon finnes ofte i hjernestammen og har svært dårligprognose, mens H3.3 G34R/V mutasjon er cortikal svulster med bedre prognose (Braunstein et al., 2017).
BRAF V600E mutasjon finnes i 5–10 % ab HGG og er forbundet med bedre prognose. For disse pasientene finnes det «target therapy» i form av BRAF inhibitor som så langt har vist lovende resultater (Robinson, Orr, & Gajjar, 2014).
Diffuse ponsgliomer
Dette er gliomer som infiltrerer diffust i den sentrale hjernestamme og utgjør en egen klinisk tilstand, uavhengig av tumors histopatologi, som i de aller fleste tilfeller er astrocytom WHO grad 2, 3 eller 4. På engelsk kalles svulsten «Diffuse Intrinsic Pontine Glioma» (DIPG). Tumor sitter nesten alltid sentralt i pons, og diagnosen stilles på bakgrunn av klassiske MR-funn og spesifikke kliniske symptomer. Prognosen er særs dårlig, bare spredte langtids overlevere er beskrevet. Det er gjort mange behandlingsforsøk med forskjellige tilnærminger uten at noen har hatt tilfredsstillende effekt. Strålebehandling har en viss livsforlengende effekt.
Det er imidlertid viktig å være klar over at det også finnes mer fokale (ikke- diffuse) svulster i hjernestammen med et helt annet forløp. Disse er som oftest lavgradige gliomer med forløp og behandling som LGG andre steder. Prognosen ved LGG i hjernestammen varierer, men kan være ganske god med høy langtidsoverlevelse. (Bemerk at fibrillært astrocytom (WHO grad 2) som manifesterer seg som DIPG ikke regnes som LGG.)
Epidemiologi
Svulster i hjernestammen utgjør 10–15 % av alle pediatriske CNS svulster hos barn og DIPG utgjør 80 % av disse. DIPG forekommer i alle aldersgrupper, men er hyppigst å alderen 5–10 år (Vanan & Eisenstat, 2015).
Patologi
Tidligere var det ikke vanlig å biopsere DIPG, både på grunn av svulstens beliggenhet i et sårbart område og begrenset terapeutiske muligheter. Muligheten for å finne molekylære endringer i svulstvevet og påfølgende målrettet terapi, har ført til en økt biopsering av DIPG, noe som har vist seg å være relativt trygt. Den pågående BIOMEDE studien for DIPG hos barn og ungdommer, krever biopsi for inklusjon i studien.
Nesten alle biopserte svulster viser astrocytom WHO grad 2–4 og fordelingen mellom disse varierer i publiserte materialer, men forløpet varierer ikke mellom de forskjellige histopatologiske typene. Det er beskrevet enkelte DIPG med PNET histopatologi.
Molekylærbiologi
Den økende biopsering ved DIPG har vist at over 80 % –90 % av DIPG innehar H3K27M mutasjon som affiserer histonfunksjon i svulstcellene (Sturm et al., 2017). Dette er bakgrunnen for den nye histologiske diagnosen i siste WHO-utgave (Gupta & Dwivedi, 2017) . Denne mutasjonen finnes også i andre høygradige midtlinjegliomer.
Diagnose og utredning
På grunn av sykdommens aggressive biologi, inntrer symptomene akutt og progredierer raskt. Oftest har symptomene vart mindre enn 2–3 måneder ved diagnose. Pasientene har klassiske hjernestammesymptomer som abducensparese, facialisparese, dysfagi, hemiplegi og ataksi.
MR cerebrum gir diagnosen. Svulsten utvider pons og infiltrerer diffust omgivelsene langs fibertraktene. Den gir betydelig masseeffekt med sammenklemming av fjerde ventrikkel, men mindre enn 10 % har hydrocefalus ved diagnose. Tumor er hypointens på T1, hyperintens på T2 og FLAIR. Den tar sjelden kontrast. Det siste er viktig differensialdiagnostisk i forhold til pilocytisk astrocytom.
DIPG diagnostiseres radiologisk og er alltid inoperabel. Biopsi av DIPG er blitt mye vanligere, grunnet muligheten for å finne molekylære endringer i svulstvevet og påfølgende målrettet terapi,
Ved ikke-diffuse hjernestammesvulster og når det er tvil om diagnosen DPG, skal det gjøres biopsi.
Behandling
Kirurgi. DIPG er alltid inoperabel. Vedrørende biopsi, se ovenfor.
Strålebehandling er standardbehandling og den eneste behandling med dokumentert effekt ved DIPG. Effekten er forbigående klinisk bedring og opptil 3 mnd. forlengelse av levetiden. Vanlig dose er 54 Gy til tumorområdet. Det gis ikke stråling til hjernen for øvrig. Behandlingen er ikke kurativ. Disse pasientene er i øyeblikket ikke kandidater for protonstrålebehandling. Re-bestråling har vist økt overlevelse, sammenliknet med standardbehandling (Lu, Welby, Mahajan, Laack, & Daniels, 2019).
Kjemoterapi og annen behandling. De fleste cytostatika i varierende kombinasjoner og doser har blitt prøvd kombinert med strålebehandling, uten bedre effekt enn strålebehandling alene. Høydosebehandling har heller ikke bedret resultatene, heller ikke Angiocomb, en studie som kombinerte topotecan som radiosensitizer gitt under strålebehandlingen, etterfulgt av kurer med de anti-angiogenetiske medikamentene thalidomid og celecoxib, samt etoposid. Utprøving med stoffer som gemcitabine (nukleosid analog), gefitinib (EGFR inhibitor), tipifarnib (farnesyltransferase inhibitor) og flere tyrosin kinase hemmere, som påvirker molekylære prosesser i svulsten, har heller ikke vist bedre resultater. I Norge (2019) inkluderer vi pasienter i BIOMEDE (Biological Medicine for Diffuse Intrinsic Pontine Glioma (DIPG) Eradication (BIOMEDE), 2014-), som er en europeisk behandlings-protokoll for DIPG. Etter biopsi fra tumor, blir alle bestrålt, i tillegg til biologisk medisin som styres etter svulstens biologiske karakter (Everlimus (mTOR inhibitor), Erlotinib eller dasatinib (begge tyrosin kinase hemmer). Resultatene fra BIOMEDE forventes i løpet av 2020.
Prognose
Prognosen er svært dårlig med median overlevelse på 9–13 måneder. Dette har ikke endret seg de senere år. Overlevelsestall varierer i litteraturen, ofte kan det være uklart om angitte overlevere egentlig har en annen type hjernestammesvulst. Det er imidlertid påvist enkeltpasienter med godt dokumentert DIPG som overlever lenge.
Det er ikke beskrevet prognostiske faktorer som har behandlingsmessige konsekvenser, men kort sykdomsvarighet og betydelige nevrologi ske symptomer har en viss negativ prognostisk betydning. Lavere malignitetsgrad (WHO grad 2) har ikke innflytelse på forløpet.
Ependymom
Ependymom er den tredje vanligste hjernesvulsten hos barn og utgjør 8–12 % av alle hjernesvulstene hos barn og ungdom. Mer enn halvparten av tilfellene opptrer hos barn under 5 år og vi antar at svulsten utgår fra ependymcellene som dekker hjernens ventrikler og sentralkanal i spinalkanalen (Khatua, Ramaswamy, & Bouffet, 2017).
Lokalisering
Ependymomene er stort sett lokalisert i tilknytning til ventriklene, men kan forekomme i hele sentralnervesystemet (CNS), uavhengig av og langt fra områder med ependymceller. Det forklares ved transformasjon av embryologiske rester av ependymceller inne i hjerneparenchymet eller en transformasjon av nevrale stamceller (Reni, Gatta, Mazza, & Vecht, 2007; Zacharoulis & Moreno, 2009). Hos barn er 90 % av ependymomene intrakranielle og av disse er 2/3 lokalisert i bakre skallegrop i tilknytning til 4. ventrikkel (Khatua et al., 2017).
Histopatologi og molekylærbiologi
Etter oppdatert WHO- klassifiseringen av CNS svulster fra 2016 skilles det mellom WHO grad I subependymom og myksopapillær ependymom, som hovedsakelig opptrer hos voksen, WHO grad II ependymom og WHO grad III anaplastisk ependymom (Gupta & Dwivedi, 2017). For WHO grad II ependymomer finnes tre histologiske varianter: pappillært, klarcellet og tancytisk ependymom. RELA fusjon positiv ependymom er en molekylær variant av supratentorielle ependymomer og kan enten være WHO grad II eller WHO grad III. De vanligste histopatologiske subtypene hos barn er klassisk og anaplastisk ependymom. Den klassiske typen viser perivaskulære pseudorosetter av gliale tumorceller uten tydelig tegn til anaplasi, mens den anaplastiske typen viser forhøyet mitotisk aktivitet, mikrovaskulær proliferasjon og utbredte nekroser. Det er glidende overganger mellom de to typene og ofte vanskelig å avgjøre hvilken gruppe de tilhører (Ellison et al., 2011). Det er foreløpig ingen pålitelige biologiske prognostiske markører for ependymom, og sammenhengen mellom det histopatologiske bildet og prognose har ikke vist seg å gjenspeile seg i kliniske studier (Ellison et al., 2011; Lundar, Due-Tonnessen, Scheie, & Brandal, 2014).
De siste årenes økning i kunnskapen om molekylærbiologiske forhold ved hjernesvulster har vist en stor heterogenitet når det gjelder ependymom og det er funnet 9 molekylære subgrupper av ependymale svulster, 3 for hvert hovedområde i hjernen: supratentorielt, bakre skallegrop og spinakanal. For supratentoriale ependymom (ST-EPN) hos barn er anaplastisk EPN med YAP1-fusjon og anaplastisk EPN med RELA-fusjon de vanlige subgruppene, hvor YAP1-fusjon er forbundet med svært god prognose og RELA-fusjon langt dårligere prognose. For ependymom i bakre skallegrop (PF-EPN) er det også hovedsakelig 2 subgrupper hos barn, anaplastisk gruppe A med dårlig prognose og anaplastisk gruppe B med god prognose. I spinalkanalen er det funnet 3 subgrupper, men alle er forholdsvis sjeldne hos barn (Pajtler et al., 2015). Forekomsten av tillegg av kromosom 1q indikerer dårlig prognose og stor sannsynlighet for tilbakefall.
Diagnose og utredning
De vanligste symptomene ved ependymom er spesifikke og relatert til økt intrakranielt trykk fra obstruktiv hydrocephalus som morgenhodepine, brekninger om morgenen og evn. personlighetsforandringer, men nevrologiske utfall og cerebellare symptomer som ataksi kan forekomme. Hos små barn kan apati og irritabilitet være de eneste symptomene. Diagnosen stilles ved MR-undersøkelse av hele CNS-aksen, undersøkelse av spinalvæsken og histologisk undersøkelse av svulstvevet.
Behandling
Behandling av ependymom er som oftest multimodal og krever høyspesialiserte, bredt sammensatte nevro-onkologiske team. Den gjennomføres i henhold til internasjonale studieprotokoller/retningslinjer.
Kirurgi
Standard behandling av ependymom er først og fremst kirurgi, etterfulgt av fokal strålebehandling (Merchant et al., 2009). Kjemoterapi har en mer usikker effekt på svulstvevet (Bouffet, Tabori, Huang, & Bartels, 2009). Den optimale behandlingen vil være total tumorfjerning, men ofte er det vanskelig grunnet svulstens evne til å infiltrere vitale strukturer, spesielt i bakre skallegrop. Målet for kirurgisk behandling vil være maksimal tumorfjerning, samtidig med et optimalt nevrologisk resultat. Studier har vist at det som betyr mest for overlevelsen, er grad av resttumor. Det har den senere tid ført til en mer aggressiv kirurgisk tilnærming med «second look» kirurgi for å oppnå totalreseksjon av tumor, og dermed bedre overlevelsen (Merchant et al., 2009). Studier har vist at det er gjennomførbart uten alvorlig morbiditet (Massimino et al., 2004).
Strålebehandling
Standard behandling av ependymom er kirurgi, etterfulgt av strålebehandling hos større barn og ungdom og kjemoterapi hos de minste barna som ikke kan få strålebehandling. Studier har vist at behandling med både kirurgi og strålebehandling har bedre overlevelse (Schroeder et al., 2008; Swanson et al., 2011) Protonstråling bør være standard behandling.
CNS-metastaser forekommer, men i langt mindre frekvens enn man tidligere trodde og derfor er det kun tumorområdet med marginer som blir bestrålt. Dosen må over 54 Gy og helst så høyt som 59.4. I den nyeste behandlingsprotokollen SIOP Ependymoma II er stråledosen 59,4 Gy for barn > 18 mnd., med mindre stråledoser (54 Gy) til barn mellom 12–18 mnd. og til barn som har gjennomgått flere kirurgiske inngrep (An International Clinical Program for the Diagnosis and Treatment of Children With Ependymoma (SIOP-EP-II), 2015-2026).
For ependymom i hjernestammen er det flere land, inkludert Norge, som har vært skeptisk til en så høy stråledose (59,4 Gy) mot et sårbart område og påfølgende fare for strålenekrose.
Cytostatika
Cytostatikabehandling ved ependymom har usikker effekt og beste standard behandling er ikke etablert (Bouffet et al., 2009). Cytostatika brukes på de minste barna i påvente av strålebehandling og cytostatika har trolig en viss effekt (Grundy et al., 2007). Andre studier viser ikke den samme effekten (Grill et al., 2001).
I «SIOP Ependymoma II» får alle spedbarn < 12 måneder og barn med resttumor, cytostatika. I protokollen inngår cyclofosfamid, vincristin, carboplatin, etoposid, cicplatin og metotrexate i forskjellige kombinasjoner. I tillegg blir spedbarn < 1 år eller barn som ikke har fått strålebehandling, randomisert til Valporat eller ikke i vedlikeholdsbehandlingen.
Barn med ependymom i Norge behandles i utgangspunktet etter «SIOP Ependymoma II», men med visse modifikasjoner. Stort sett vil en i Norge gå for mindre stråling enn anbefalt i protokollen når tumor ligger i hjernestammen. Faggruppen for CNS svulster hos barn har bestemt at Norge skal delta i «SIOP Ependymoma II», og arbeidet med å åpne studien i Norge er allerede i gang.
Prognose
Prognosen for ependymom er relativt dårlig med 5 års PFS mellom 23–74 % og OS mellom 60 %–80 % (Cage et al., 2013). Den varierer med alder, WHO-klassifikasjon, lokalisasjon og resttumor etter kirurgi. Det som har betydd mest prognostisk er grad av resttumor etter kirurgi og ved komplett reseksjon av WHO grad 1–3 er det > 60 % 5-års overlevelse (Pajtler et al., 2017; Pejavar et al., 2012). Overlevelse basert på de nye molekylære subgruppene av ependymale svulster, viser at ST anaplastisk EPN med YAP1-fusjon og PF anaplastisk EPN gruppe B har svært god prognose (10 års OS 88–100 %), mens ST anaplastisk RELA-fusjon og PF anaplastisk EPN gruppe A har langt dårligere prognose (10 års OS 50 %) (Pajtler et al., 2015).
Oppfølging
Alle barn kontrolleres med klinisk undersøkelse og MR av hele CNS hver 3. mnd. første året etter avsluttet behandling, deretter hver 4. mnd. andre året og videre halvårige kontroller frem til 5 år etter avsluttet behandling. I tillegg følges alle barn minst 1 gang årlig med tanke på sekveler av behandlingen. Mange barn med ependymom vil i varierende grad få senfølger av behandlingen. Dette må følges nøye. Nevropsykologisk kartlegging gjennomføres på alle 1, 2 og 5 år etter diagnose i hht nasjonalt tverrfaglig oppfølgingsprogram, og det gjøres en vurdering av behov for dette også tidligere i forløpet. Pedagogisk oppfølging ved behov er vesentlig. Videre oppfølging av barnenevrolog, fysioterapeut, endokrinolog, nyrefunksjon, hørsel, ØNH-status og øyestatus gjøres i varierende grad. Der bivirkninger av behandlingen påvises, etableres det et samarbeid med lokale aktører for å legge til rette for best mulig habilitering
Residivbehandling
Residiv ved ependymom forekommer hos 30–50 % av pasientene og er forbundet med veldig dårlig prognose (Zacharoulis et al., 2010). Det finnes ingen standard behandling. Nytt operativt inngrep må alltid vurderes og der hvor strålebehandling ikke er gjennomført, må det vurderes. Kraniospinal re-bestråling har vist økt overlevelse sammenliknet med fokal re-strålebehandling, mens cytostatika har vist dårlig effekt (Tsang, Murray, et al., 2019). Utprøvende behandling med biologisk medisin vil mulig gi større håp for denne pasientgruppen i fremtiden.
Medulloblastom
Medulloblastom er en malign svulst av embryonal opprinnelse lokalisert i lillehjernen, som opptrer særlig hos barn. Det er en biologisk heterogen svulstgruppe, og det er nå bred enighet om at det eksisterer 4 molekylærgenetiske subgrupper med betydning for prognose. I WHOs klassifikasjon av svulster i sentralnervesystemet fra 2016 er disse nå tatt med, i tillegg til de histologiske undergruppene av medulloblastom (Louis et al., 2016).
Symptomene er enten sekundært til svulstens tendens til å gi vannhode/ hydrocephalus eller direkte påvirkning av (lille)hjerne eller ryggmarg. Kvalme, brekninger, trunkal ataxi og koordinasjonsproblemer er de vanligste symptomene.
Diagnose
MR-undersøkelse som fremstiller hjerne og ryggmarg er nødvendig og gir mistanke om diagnosen, men endelig diagnose og klassifisering kan ikke bestemmes før etter histologisk og molekylærbiologisk undersøkelse av svulstmaterialet og spinalvæske.
WHOs klassifikasjon av medulloblastomer (2016)
Medulloblastomer klassifiseres under embryonale tumores og er høygradig maligne (WHO grad IV). Følgende undergrupper er definert:
Både molekylærbiologisk og histologisk definert medulloblastom brukes når diagnosen stilles, og de ulike kombinasjonene av genetiske og histologiske undergrupper har ulik prognose og forekommer i ulike aldersgrupper. Se tabell under:
Table 5: Summary of the most common integrated medulloblastoma diagnoses, with clinical correlates
Molekylærbiologiske markører
Tumormateriale vil bli undersøkt på følgende markører for å evaluere prognostisk betydning: CTNNB1-mutasjoner (WNT-aktiverte medulloblastomer), APC-mutasjoner (WNT-aktiverte CTNNB1-villtype medulloblastomer), monosomi 6 (WNT-aktiverte medulloblastomer), gain/amplifikasjon av C-MYC og N-MYC, TP53-mutasjoner, PTCH1-mutasjoner, og SUFU-mutasjoner (SHH-aktiverte medulloblastomer).
Flere av disse markørene er implementert som prognostiske faktorer i behandlingsprotokoller.
Stadieinndeling (Chang)
M0: | Lokalisert til lillehjernen uten tegn til lokal spredning eller spredning til hjernehinnevæsken |
M1: | Spredning til hjernehinnevæsken |
M2: | Spredning lokalt i lillehjernen |
M3: | Spredning til storehjerne/ryggmargen |
M4: | Spredning utenfor sentralnervesystemet |
Standard risk: | M0 og postoperativ resttumor under 1.5 cm2 |
Høyrisk: | M1–4, storcellet eller anaplastisk histologi, og/eller MYC-amplifisert |
Behandling
Kirurgi alene er ikke kurativt ved medulloblastom. Adjuvant behandling er helt nødvendig for overlevelse. Barn over 4 år vil rutinemessig bli behandlet med strålebehandling, helst protonbestråling, mot hjerne og ryggmarg. Barn under 4 år vil bli behandlet med kjemoterapi som eneste adjuvante behandling. Opptil 1/3 av pasientene har spredning til hjernehinnene ved diagnose. Valg av behandlingsprotokoll og -stratifisering vurderes og bestemmes i MDT-møte.
Kirurgi er meget viktig for prognosen da minimal restsykdom etter kirurgi gir best prognose. Ved restsvulst på mindre enn 1,5 cm2 på postoperative MR-bilder ser man en bedre prognose/overlevelse enn ved større resttumor. Som for de fleste andre svulsttyper vil derfor målsetningen med kirurgi ved medulloblastom være maksimal svulstreseksjon uten å løpe for stor risiko for å påføre pasienten nevrologiske sekveler.
Stråleterapi er en viktig del av behandlingen og hele nevralaksen skal bestråles med boost mot primærtumorområdet og eventuelle metastaser. Dose til nevralaksen varierer fra 18 til ca 36 Gy, mens man til primærtumorområdet og eventuelle metastaser totalt gir 54 Gy (ca 50 Gy ved bestråling spinalt). Grunnet risiko for alvorlige senskader anbefales ikke bestråling til de minste barna. Stråling med protoner er standardbehandling i dag. Mer spesifikt gir man for norske barn under 4 år ikke strålebehandling til nevralaksen, mens man vurderer om det er aktuelt med lokal bestråling mot tumortomt og eventuell resttumor for de som er fra 2 år og oppover. Ett av ankepunktene mot sistnevnte strategi er at det ved recidiv blir vanskeligere å gi ny strålebehandling.
Cytostatikabehandling er under kontinuerlig evaluering. Medulloblastomer er vanligvis kjemoterapisensitive, men beste standardbehandling er ikke endelig fastslått. Hos barn < 4 år startes det med postoperativ cytostatikabehandling i håp om å utsette stråleterapi lengst mulig (Rutkowski, 2006; Rutkowski et al., 2005). Hos noen spesifikke undergrupper av medulloblastom hos de aller yngste barna, er det også god prognose ved kun adjuvant behandling med cytostatika, inkludert methotrexate intratekalt.
Standard risk < 4 år: | HIT MED Guidance |
Standard risk > 4 år: | SIOP PNET 5 |
Høyrisk alle aldre: | HIT MED Guidance |
Cytostatika som inngår er cyclofosfamid, vinkristin, metotrexat, karboplatin, etoposid, cisplatin og CCNU i varierende kombinasjoner og intensitet alt etter alder og sykdomsutbredelse. Mange av høyrisk-pasientene og de under 4 år får også intratekal metotrexat. Alle barn over 4 år får bestråling mot CNS-akse. Barn under 4 år behandles rutinemessig med kun cytostatika.
Prognose
Best prognose har standard risk pasienter med ca 70 % langtidsoverlevelse, mens prognosen for de yngste barna (< 4 år) er dårligere. Høyriskpasientene > 4 år har ca 40–50 % langtidsoverlevelse, og også blant disse er det dårligst prognose for de minste barna (< 4 år).
Oppfølging
Alle barn kontrolleres og diskuteres i MDT-møter. Rutinemessig billedoppdatering og tumorkontroll etter 3, 6, 12 18 og 24 mnd med MR, siden MR hvert år til 5 år etter avsluttet behandling. Etter 5 år er det individuell oppfølging, men pga at de fleste har fått strålebehandling mot total CNS-akse, vil vi anbefale MR-kontroller livslangt.
I tillegg bør alle barn følges minst 1 gang årlig med tanke på sekveler av behandlingen. Alle barn med medulloblastom vil i varierende grad få senfølger av behandlingen. Nevropsykologisk kartlegging gjennomføres for alle 1, 2 og 5 år etter diagnosetidspunkt i hht nasjonalt tverrfaglig oppfølgingprogram for barn med tumor cerebri og det gjøres en vurdering av behov for dette også tidligere i forløpet. Pedagogisk oppfølging ved behov er vesentlig. Videre oppfølging av barnenevrolog, fysioterapeut, endokrinolog, nyrefunksjon, hørsel, ØNH-status og øyestatus gjøres i varierende grad. Der bivirkninger av behandlingen påvises, etableres det et samarbeid med lokale aktører for å legge til rette best mulig habilitering.
Residivbehandling
Ved residiv er behandlingsresultatene nedslående. De terapeutiske mulighetene er i de fleste tilfellene brukt i første omgang. Der hvor strålebehandling ikke er gjennomført, må det nå vurderes. Reoperasjon kan være aktuelt. Førstevalget i Norge for barn med residiv av medulloblastom, er Memmat-protokollen. Denne protokollen er nå åpen for inklusjon av norske pasienter. Dette er en metronomisk og målrettet anti-angiogenetisk behandlingsprotokoll, som også inkluderer intratekal cellegift.
Supratentoriell PNET
Andre embryonale hjernesvulster
Disse svulstene gikk tidligere under fellesbetegnelsen supratentoriell PNET (primitiv nevroektodermal tumor). Medulloblastom er en primitiv nevroektodermal tumor lokalisert i lillehjernen, mens disse andre ble kalt supratentoriell PNET da de var lokalisert supratentorielt. PNET er imidlertid ikke lenger en egen gruppe svulster i WHO-klassifiseringen fra 2016, men er nå delt opp i flere ulike entiteter i gruppen embyonale svulster (Louis et al., 2016).
Uspesifikke symptomer som tegn på økt intrakranielt trykk, kramper og nevrologiske utfall er det som oftest bringer pasienten til kontakt med helsevesenet.
Diagnosen stilles ved MR-undersøkelse. Hele CNS-aksen må visualiseres og vevsprøve fra svulsten samt cytologi fra spinalvæsken må undersøkes for endelig diagnose og klassifisering.
WHOs klassifikasjon av embryonale svulster som ikke er medulloblastomee (2016) inneholder følgende undergrupper:
- Embryonal tumor med rosetter, C19MC-endret (ETMR)
- Embryonal tumor med rosetter, NOS (ETMR)
- Medulloepiteliom
- CNS nevroblastom
- CNS ganglionevroblastom
- CNS embryonal svulst, NOS
- Atypisk teratoid/rhabdoid tumor (se eget kapittel)
- CNS embryonal tumor med rhabdoide trekk
Molekylærbiologiske markører
Tumormateriale vil bli undersøkt med tanke på mange molekykærbiologiske markører, noe avhengig av hvilken type embryonal svulst man mistenker. For eksempel vil CNS nevroblastom oftest ha gevinst av kromosomarm 1p, tap av kromosomarm 16q og FOXR2-forandringer,
Behandling
Behandling av disse embryonale svulstene krever høyspesialiserte og bredt sammensatte nevroonkologiske team (MDT). Behandlingen gjennomføres i henhold til internasjonale studieprotokoller/retningslinjer (B. H. Cohen et al., 1995; Massimino et al., 2006; Timmermann et al., 2006).
Kirurgi er meget viktig for prognosen da minimal restsykdom etter kirurgi gir best prognose. Operasjoner bør derfor gjøres der det finnes erfarne nevrokirurger med omfattende erfaring med barn og god barneanestesi-/intensivekspertise.
Stråleterapi er en viktig del av behandlingen og for å ha et realistisk håp om kurasjon må hele nevralaksen bestråles med boost mot primærtumorområdet og eventuelle metastaser. Dose til nevralaksen vil for de fleste være ca 36 Gy, mens man til primærtumorområdet og eventuelle metastaser totalt gir 54 Gy (ca 50 Gy ved bestråling spinalt). Grunnet risiko for alvorlige senskader anbefales ikke bestråling til de minste barna (Timmermann et al., 2006). Mer spesifikt gir man for norske barn under 4 år ikke strålebehandling til nevralaksen, mens man vurderer om det er aktuelt med lokal bestråling mot tumortomt og eventuell resttumor for de som er fra 2 år og oppover. Ett av ankepunktene mot sistnevnte strategi er at det ved recidiv blir vanskeligere å gi ny strålebehandling. Stråling med protoner bør alltid vurderes.
Cytostatikabehandling er under kontinuerlig evaluering. Pasientene behandles oftest i henhold til HIT MED Guidance. Hos barn < 4 år startes det med postoperativ cytostatikabehandling i håp om å utsette stråleterapi lengst mulig.
Prognose
Prognosen varierer avhengig med type embryonal svulst og er vesentlig dårligere enn for medulloblastom. For hele gruppen (som tidligere ble benevnt PNET) ligger langtidsoverlevelsen ligger på ca 30–40 % (B. H. Cohen et al., 1995).
Oppfølging
Alle barn kontrolleres for tumorstatus etter 3, 6, 12 18 og 24 mnd med MR, siden MR hvert år til 5 år etter avsluttet behandling. Etter 5 år er det individuell oppfølging, men pga at de fleste har fått strålebehandling mot total CNS-akse, vil vi anbefale MR-kontroller livslangt.
I tillegg følges alle barn minst 1 gang årlig med tanke på sekveler av behandlingen. I varierende grad gjøres nevropsykologisk og spesialpedagogisk testing, undersøkelse av barnenevrolog, fysioterapeut, endokrinolog, nyrefunksjon, hørsel, ØNH-status og øyestatus. Der bivirkninger av behandlingen påvises, etableres det et samarbeid med lokale aktører for å legge til rette best mulig habilitering. Alle barn med PNET vil i varierende grad får senfølger av behandlingen som må følges nøye, og spesielt må endokrinologisk substitusjonsbehandling og pedagogisk tilrettelegging etableres.
Residivbehandling
Residivbehandling er nedslående. Der hvor strålebehandling tidligere ikke er gjennomført, må det nå vurderes. Reoperasjon kan være aktuelt. Utprøvende behandling med cytostatika kan startes, men resultatene så langt er ikke oppløftende.
Atypisk teratoid-rhabdoid tumor/ATRT
Atypisk teratoid / rhabdoid tumor beliggende i CNS (CNS AT/RT) er en svært ondartet svulst som hovedsakelig forekommer hos barn under tre år.
Diagnosen stilles ved histologisk undersøkelse.
Billedundersøkelse: Hele CNS-aksen må visualiseres og spinalvæsken undersøkes for klassifisering og utbredelse. Billedundersøkelse viser ofte cyster eller blødninger. På T1-vektet MR er svulstene hypointense, mens T2-vektede bilder viser iso- til hyperintense-lesjoner. Det er heterogen kontrastladning og ved leptomeningeal sykdom ser man ved billedundersøkelse en nodulær, klumpete kontrastoppladning langs meningene i spinalkanalen og inn i cauda equina. Billedundersøkelse alene kan ikke sikkert skille mellom AT/RT og suptratentorielle nevroblastomer (tidligere PNET) eller medulloblastom. Omtrent to tredjedeler av AT/RT-er forekommer i lillehjernen, vanligvis i den cerebellopontinvinkel, med infiltrasjon av omgivende strukturer. Omtrent en fjerdedel er supratentorial og 8 prosent er multifokale.
Patologi: Histologisk er AT/RT preget av rhabdoide celler og ligner på andre små runde blåcelletumorer. AT/RT kan imitere flere forskjellige entiteter; også medulloblastomer Nekrose og en høy grad av mitotisk aktivitet er vanlig. Germcellemarkører er negative.
Biologiske markører: Diagnostisering av AT/RT krever tap av enten INI1 kjernefarging, noe som indikerer biallel inaktivering av SMARCB1 på kromosom 22, eller tap av BRG1-farging, noe som indikerer SMARCA4 inaktivering. I tillegg til disse somatiske mutasjonene påvist i tumorvev, har omtrent en tredjedel av pasientene kimcelle mutasjoner i SMARCB1 eller, mindre vanlig, SMARCA4. Vanligvis er de minst delvis vimentin, CK, EMA og smooth muscle actin (SMA) positive. Ki67 er vanligvis relativt høy. Med hjelp av OTX2 fargning er det mulig å undersøke om det dreier om en AT/RT-Tyr. /flere av disse markørene vil kunne bli implementert som prognostiske faktorer i fremtidig protokoller (Japp, Klein-Hitpass, Denkhaus, & Pietsch, 2017; Johann et al., 2016).
Epidemiologi
AT/RT er den mest vanlige ondartede CNS tumores hos barn under 1 år, og AT/RT representerer omtrent 1–2 % hjernesvulster hos barn. I Norge har det vært 19 tilfeller av AT/RT i de 15 årene 2002–2017, det tilsier i snitt drøyt en pasient i året i hele Norge.
Symptomer: Symptomer hos spedbarn er ofte slapphet, oppkast og eller økende hodeomkrets; eldre barn kan ha symptomer grunnet affeksjon av hjernenervene IV, VI eller VII, for eksempel torticollis, dobbeltsyn eller facialisparese. Hemiplegi og/eller hodepine generelle trykksymptomer kan også være debutsymptomer.
Prognosen: I de fleste studier og rapporter er dårlig, med en median overlevelse på 12 til 14 måneder og fem års samlet overlevelse mindre enn 50 prosent. Pasienter med familiemutasjoner i SMARCB1genet har en tendens til å være yngre og ha dårligere prognose sammenlignet med de med ikke familiære, sporadiske svulster. Faktorer som er assosiert med bedre prognose inkluderer supratentoriell beliggenhet, ikke spredning av sykdommen ved diagnosetidspunktet, fullstendig reseksjon av tumor kirurgisk og uttrykk for ASCL1, en regulator for NOTCH-signalering.
Behandling: Kirurgi alene er ikke kurativt ved AT/RT, men en kombinasjon av kirurgi, omfattende cellegiftbehandling og strålebehandling er helt nødvendig for overlevelse (Fossey et al., 2017; Richardson, Ho, & Huang, 2018; Schrey et al., 2016).
Kirurgi er meget viktig for prognosen da minimal restsykdom etter kirurgi gir best prognose. Målsetningen med kirurgi ved AT/RT som ved andre ondartede CNS svulster er maksimal reseksjon.
Cytostatikabehandling er under kontinuerlig evaluering, og til nå har vi fulgt en anbefaling gitt i «A multinational registry for rhabdoid tumors of any anatomical site EUROPEAN RHABDOID REGISTRY, EU-RHA». AT/RT registeret er en anbefaling både når det gjelder utredning, behandling og oppfølgning av AT/RT. Her anbefales det å gi 9 kurer med kjemoterapi hver 2. uke, dels intravenøst og dels også intrathekalt i Ommaya reservoir. AT/RT svulstene er vanligvis initialt kjemosensitive. Alle barn får cytostatika under strålebehandling, men kurene modifiseres under strålebehandlingen på grunn av fare for økt toksisitet. I anbefalingene som er gitt i dette registret, har man også foreslått høydosebehandling med stamcellestøtte. Til nå har ikke høydosebehandling vist noen signifikant bedret overlevelse enn de 9 regulære kjemoterapikurer. Etter strålebehandling skal man ikke lenger gi intrathekal kjemoterapi.
Stråleterapi er en viktig del av behandlingen for å ha et realistisk håp om kurasjon, og man anbefaler å komme i gang med strålebehandlingen så tidlig som mulig i forløpet, i løpet av de første kurene. Ved ung alder og ikke dissiminert sykdom, anbefales lokal stråling mot tumorsengen, men ved eldre alder, resttumor og disseminert sykdom, anbefales å stråle hele nevralaksen med boost mot primærtumorområdet og eventuelle metastaser. Grunnet risiko for alvorlige senskader anbefales ikke CNS akse bestråling til de minste barna. Man anbefaler stråling med protoner.
Videre studier/planlagt behandling: Det er en planlagt protokoll med stratifiseringer og randomiseringer på trappene, men den kommende AT/RT protokoll er ikke offisiell ennå. Den vesentlige endringen er å gi 12 runder kjemoterpi i stedet for 9, : 3 dox, 4 VCA og 5 ICE.
Oppfølging: Alle barn kontrolleres med tanke på tumorkontroll etter 3, 6, 12 18 og 24 mnd med MR, deretter MR minimum to ganger årlig fra 2 til 5 år etter avsluttet behandling, og årlig til 10 år etter ferdig behandling. De barna som ikke har fått strålebehandling, anbefales å ta spinalpunksjon 2 ganger i året i 2 år etter ferdig behandling.
I tillegg følges alle barn minst 1 gang årlig med tanke på sekveler av behandlingen. Alle barn med AT/RT vil i varierende grad få senfølger av behandlingen som må følges nøye. Nevropsykologisk testing gjennomføres 1, 2 og 5 år etter diagnose i hht nasjonalt tverrfaglig oppfølgingprofgram for barn med tumor cerebri og det gjøres en vurdering av behov også tidligere i forløpet. Pedagogisk oppfølging ved behov er vesentlig. I varierende grad får barnet oppfølging av barnenevrolog, fysioterapeut, endokrinolog, nyrefunksjon, hørsel, ØNH-status og øyestatus. Det er en målsetting at der bivirkninger av behandlingen påvises, etableres samarbeid med lokale aktører for å legge til rette best mulig habilitering.
Residivbehandling: Ved residiv er behandlingsresultatene nedslående. De terapeutiske mulighetene er i de fleste tilfellene brukt i første omgang. Der hvor strålebehandling ikke er gjennomført, må det nå vurderes. Reoperasjon er sjelden aktuelt, da residivet ofte viser rask vekst, og tumor ikke lar seg fjerne. MEMMAT protokoll med metronomisk behandling med blant annet Avastin, peroral kjemoterapi, intrathekal behandling og diverse perorale medisiner (Celecoxib, Fenofibrat, Taidomid) kan vurderes. Målrettede terapier er også under utredning i en rekke fase-1 studier, og et stort antall medikamenter har potensiell terapeutisk aktivitet. Spesielt Aurora kinase A-hemmeren Alisertib, (MLN8237) har vist antitumoraktivitet hos barn med tilbakefall av AT/RT eller resistent sykdom.
Germinalcellesvulster i CNS
Germinalcelletumores (GCT) er en gruppe sjeldne svulster med antatt utgangspunkt i totipotente primordiale germinalceller, som migrerer til «feil plass» og kan undergå malign transformasjon i løpet av vekst og differensiering. De er heterogene både mht primærlokalisasjon, histologi, biologisk profil og behandlingsrespons. Totalt utgjør GCT ca. 3,4 % av maligne svulster hos barn, og ca. 1/5 av dem oppstår i sentralnervesystemet. Insidenskurven er to‑puklet, høyest hos sped-/småbarn med en ny stigning rundt puberteten, og de er hyppigere hos gutter
Lokalisasjon
De finnes oftest i midtlinjen, selv om andre lokalisasjoner forekommer. Særlig vanlig er pinealregionen. Undergruppen «germinomer» kan være bifokale (pinealregion + suprasellært) uten at dette regnes som metastatisk sykdom. Vanligste spredningsform er via utsæd i cerebrospinalvæsken.
Symptomer/tegn
Avhenger av lokalisasjon. Ved beliggenhet i pinealregionen oppstår ofte hydrocephalus med trykksymptomer pga aqueductstenose. Videre sees ofte øyepareser og diplopi, samt «Parinauds syndrom» (blikkparese oppad og dissossiert pupillerefleks med intakt akkomodasjon, men manglende lysrefleks). Ved suprasellær lokalisasjon omfatter symptombildet foruten synsforstyrrelser også ofte diabetes insipidus og andre endokrine dysfunksjoner, som f.eks. forsinket pubertet.
Diagnose
Ved mistanke om intracerebral GCT bør pasienten utredes videre ved spesialavdeling. MR-undersøkelsen må omfatte hele CNS-aksen i henhold til spesifiserte retningslinjer i SIOP CNS GCT II-protokollen. Spinalvæsken skal undersøkes cytologisk for utsæd av tumorceller. Det må gjøres før tumor er manipulert operativt. Dersom det har vært behov for ø.hj.-operasjon pga høyt intracranielt trykk, bør det gå minst 10 dager før undersøkelsen foretas. Tumormarkørene AFP og Beta-HCG skal analyseres i både serum og cerebrospinalvæske. Dersom disse er forhøyet (i én eller begge komponenter), kan man fastslå at det dreier seg om en «secreting germ cell tumor» i undergruppen malignt non-germinom (NG-GCT). Dermed kan biopsi unngås. Men dersom markørene er under de definerte grenseverdiene kreves histologisk verifikasjon, dog med unntak av bifocal sykdom med karakteristisk lokalisasjon, som er typisk for germinom (se ovenfor).
Utredningen må omfatte så vel utbredelsesgrad som histologisk subtype, som skissert i følgende figurerer fra SIOP-CNS GCT II-protokollen.
Rekkefølgen av undersøkelsene avhenger av om det foreligger behandlingstrengende akutte symptomer:
Patologi/Histologi
Germinalcellesvulster kan fremvise:
- Germinalcelle differensiering (→germinom, som tilsvarer seminom og dysgerminom i testis)
- Somatisk diffensiering:
- Embryonal differensiering (→ embryonalt carcinom)
- Ekstraembryonal (→choriocarcinom og → plommesekktumor = yolksac tumor = YST)
- Teratom (moden/umoden) med tilstedeværelse av modne eller umodne strukturer fra alle 3 germinal-lag (graderes 1–3 avhengig av relativ mengde umodent vev).
NB: Ofte forekommer blandingsformer!
Histologien hos nyfødte er nesten alltid teratom (modne og umodne), i blant med små foci av plommesekktumor. I spedbarns- og småbarnsalder dominerer YST, mens andel germinomer stiger i senere barne- og ungdomsår.
Tumormarkørene har tendens til overlapping mellom undergruppene, teratomceller kan f.eks. være noe positive for AFP. Det er avgjørende viktig for valg av behandling å undersøke for et bredt immunhistokjemisk/molekylærbiologisk/cytogenetisk panel.
Histologi | Klinikk | Tumormarkører | Følsom for | ||
|---|---|---|---|---|---|
|
| AFP | Tot. HCG | Cytostatika | RT |
Germinom | malign | – | (+) | +++ | +++ |
Malignt non-germinom (secreting GCT): |
|
|
|
|
|
Embryonalt carcinom | malign | – | – | +++ | ++ |
Plommesekktumor | malign | +++ | – | +++ | ++ |
Choriocarcinom | malign | – | +++ | +++ | ++ |
Teratom | benign (potensielt malign) | – | – | –/? | +/– |
Behandling
Både utredning og behandling av GCTs krever nevroonkologiske spesialistteam, og pasienten må snarest mulig henvises til et universitetssykehus.
Prinsippet for valg av terapi ved sammensatte GCTs er at man skal behandle i henhold til den mest maligne komponenten. Dersom full utredning mht markører og utsæd ikke er foretatt, må sykdommen behandles som metastatisk sykdom av den mest maligne undergruppe.
Det er meget viktig å sikre en fullverdig utredning nært opptil oppstart av behandling (min. 14 dager før, ellers kreves ny utredning).
Protokollen SIOP CNS GCT II er stengt for inkludering av nye pasienter, men brukes fortsatt som utgangspunkt for behandlingen av maligne germinalcellesvulster. En ny protokoll er under utarbeidelse, men avhenger av resultatene av nåværende protokoll (analyseres etter follow up avsluttes i 2020).
Generelt avhenger behandlingsintensiviteten av histologi, utbredelsesgrad, AFP-nivå og alder (grense 6 år). Strategien må korrigeres avhengig av evalueringer underveis, bl.a. med henblikk på behandlingsrespons.
Nevrokirurgi er viktig for biopsi der dette er påkrevet, samt ved behov for trykkavløsende ventriculostomi. Ved rene teratomer er kirurgi hovedbehandlingen. Kirurgisk reseksjon kan også være aktuelt ved resttumor etter kjemoterapi for andre histologiske undergrupper.
Teratom behandles i hovedsak med kirurgi. Evt. tilleggsbehandling individualiseres etter histologi, reseksjonsstatus mv. Studiesentret kan kontaktes for råd.
Lokalisert germinom behandles med kjemoterapi og ventrikulær strålebehandling med fokal boost, uansett om de er i komplett eller partiell remisjon etter kjemoterapi (ihht partiell remisjon etter kjemoterapi i protokollen (standard behandling)).
Germinom med metastaser behandles med strålebehandling mot total-CNS (CSI) ihht SIOP CNS GCT II-protokollen.
Lokalisert NG-GCT behandles med kjemoterapi og strålebehandling. Kjemoterapien er PEI-kurer (Cisplatin, Etoposid og Ifosfamid) ihht standard risiko pasienter i protokollen. For høy-risiko NG-GCT (AFP>1000 ng/ml el. diagnosealder < 6år) diskuteres valg av kjemoterapi. Studiesentret kan kontaktes for råd. Strålebehandlingen er lokal ihht SIOP CNS GCT II-protokollen, men inkludering av ventriklene vurderes, spesielt gjelder dette ved protonbestråling.
NG-GCT med metastaser behandles med kjemoterapi og strålebehandling. Kjemoterapien er PEI-kurer (Cisplatin, Etoposid og Ifosfamid) ihht standard risiko pasienter i protokollen. For høy-risiko NG-GCT (AFP>1000 ng/ml el. diagnosealder < 6år) diskuteres valg av kjemoterapi. Studiesentret kan kontaktes for råd. Strålebehandlingen mot total-CNS (CSI) ihht SIOP CNS GCT II-protokollen.
Prognose
Best prognose har germinom, med 90–95 % 5-års EFS i foregående SIOP-protokoll (CNS GCT-96) som inneværende bygger videre på. Ved NG-GCT er EFS rundt 70 %.
Oppfølging
Detaljert oppfølgningsplan både m.h.t residiv og senskader foreligger i protokollen. Residiv ved maligne GCTs er hyppigst de første 2 år etter behandling. Alle pasienter skal følges opp (inkl. klinikk, MR og markører) hver 3. måned første år og videre i h.h.t. protokoll minst 5 år etter avsluttet behandling. Ved germinom (som kan residivere enda senere) anbefales oppfølgning minst 10 år.
Barn med germinalcelletumor vil i varierende grad få senfølger av behandlingen som må følges nøye. Nevropsykologisk testing gjennomføres 1, 2 og 5 år etter diagnose i hht nasjonalt tverrfaglig oppfølgingprogram for barn med tumor cerebri, og det gjøres også en vurdering av behov for dette tidligere i forløpet. Pedagogisk oppfølging ved behov er vesentlig. I varierende grad får barnet oppfølging av barnenevrolog, fysioterapeut, endokrinolog, nyrefunksjon, hørsel, ØNH-status og øyestatus. Det er en målsetting at der bivirkninger av behandlingen påvises, etableres samarbeid med lokale aktører for å legge til rette best mulig habilitering.
For mer informasjon, se referanselisten til «SIOP CNS GCT II protocol for the diagnosis and treatment of children, adolescents and young adults with intracranial Germ Cell Tumours» (Prospective Trial for the Diagnosis and Treatment of Intracranial Germ Cell Tumors (SIOPCNSGCTII), 2011-), hvor erfaringer fra en lang rekke europeiske, amerikanske og japanske studier er omtalt (Fullstendig protokoll tilgjengelig på hjemmesidene til KSSB).
Kraniofaryngeom
Kraniofaryngeom er en histologisk benign epitelial svulst utgående fra embryonale rester av ductus craniopharyngeus.
På tross av benign histologi fører slike svulster hos mange pasienter til betydelige senskader i form av hypopituitarisme, massiv overvekt, synsforstyrrelser og kognitive skader. Dette skyldes beliggenheten i hypothalamus og nærhet til synsveiene.
Kraniofaryngeomer klassifiseres til WHO grad I.
Etiologien er ukjent og det foreligger ingen kjente genetiske disposisjoner.
Forekomst
Kraniofaryngeomene utgjør ca. 3 % av intrakranielle svulster hos barn, dvs 1–2 nye tilfeller i Norge pr år. Ingen kjønnsforskjell.
Det er to ulike manifestasjoner ved vevsundersøkelse, adamantinomatøs type som er helt dominerende hos barn, mens voksne oftest har papillær type.
Molekylærbiologi
Det er nylig påvist at barnekraniofaryngeom, den adamantinomatøse typen, har en mutasjon i genet til beta-catenin som er viktig for wnt-signalveien. Mutasjonen fører til overaktivering av beta-catenin og derved wnt. Dette aktiverer også andre signalveier «downstream» i påvirkningsrekken med tumorutvikling.
Lokalisasjon
Vanligst suprasellært, evt med intrasellær komponent. Kan være rent intrasellær, mens enkelte har svær, gjerne cystisk utbredelse, også til 3. ventrikkel. Ektopisk lokalisasjon (ekstradural) forekommer.
Symptomer
Synsaffeksjon som uttrykk for kompresjon på synsbanekrysningen og synsbaner. Vekst i hypothalamus og hypofyse gir hormonelle forstyrrelser, herunder ikke sjelden vekstforstyrrelser.
Noen debuterer med diabetes insipidus. Hypothalamuslesjon kan gi kraftig overvekt og også kognitiv svikt. Ved store svulster økt intracranielt trykk evt med hydrocephalus.
Diagnose og utredning
MR undersøkelser gir vanligvis avklarende diagnostikk. CT kan vise kalk. Ved stor cyste tappes vanligvis fra denne med funn av «maskinoljelignende» væske. Biopsi gir sikker diagnose.
Hormonutredning av hypothalamus-hypofyseaksen og diabetes insipidus er viktig. Likeledes synsundersøking, vekstforhold og kognitiv status.
Behandling
Internasjonalt har man ikke kommet frem til en klar og entydig algoritme for behandling av denne vanskelig tumortypen (Muller, 2010). Hvert enkelt barn må vurderes for et individualisert behandlingstilbud basert på vurderinger ut fra pasientens alder, kliniske tilstand og tumors størrelse og vekstmønster.
Kirurgi har tradisjonelt vært primær behandling hvor man har tilstrebet radikalitet. Det er imidlertid en økende internasjonal trend å være skånsom ved reseksjon for å unngå skade på hypothalamus (Puget et al., 2007). Hypothalamuslesjon er en følge av tumors infiltrasjon, men det har vist seg at ved kirurgi i denne regionen kan sekvelene øke betydelig. En tilstreber derfor kirurgi av tumordelen utenfor hypothalamus, ikke minst hvis det er påvirkning av synsveiene. Biopsi gjøres ved diagnose.
Ved inkomplett reseksjon kan postoperativ strålebehandling / stråleknivbehandling være aktuelt for å redusere risiko for videre vekst. En større tysk studie viser at en ikke taper noe ved å utsette en slik behandling inntil det har inntruffet vekst av tumor etter kirurgien.
Intracystisk behandling. Hos barn er de fleste svulstene cystiske og ved store cyster kan man initialt implantere kateter for tømming av cystene og avlasting av press på omgivelsene. Instillasjon i cystene av cellegift (bleomycin) eller isotop (Yttrium) har tidligere vært brukt i behandlingen. Komplikasjonene har imidlertid vært utfordrende og behandlingen komplisert. I de senere år har instillasjon av interferon blitt standardbehandling ved flere sentra. Man oppnår ikke radikal fjerning av tumor, men avlastning av synsnerver og omkringliggende vev med stor grad av symptomfrihet. Pasienten må følges med regelmessige billedoppdateringer til nydanning av cystene opphører og hvis det ikke er mulig å kontrollere cystene med tapping/ interferon kan det være indisert med annen kirurgisk behandling evt i kombinasjon med stråleterapi.
Systemisk medikamentell behandling er i dag på forsøksstadiet. Signalveier «downstream» for wnt-signalveien aktiveres ved kraniofaryngeom, for eksempel MAPK-signalveien (RAS/RAF/MEK/ERK). En har derfor nå i England startet forsøk med MEK-inhibitor i utvalgte tilfeller. Resultater foreligger ikke nå.
En annen tilnærming er antiinflammatorisk behandling, da inflammasjon er en viktig del av patogenesen ved disse svulstene. Aktuelt er Interleukin-1 Reseptor-inhibitor (anakinra). Kliniske forsøk vil komme.
Symptomatisk behandling: Det er vesentlig at barn med slike svulster følges opp i et multidisiplinært team (MDT) med bred erfaring innen nevrokirurgi, nevroonkologi og nevroendokrinologi. Hormonutfall må monitoreres og substitueres. Vektøkning må prøves begrenset Videre er det viktig med oppfølging av kognitive funksjoner, synsfunksjon osv.
Prognose
Selv etter radiologisk tilsynelatende radikal kirurgisk reseksjon ser man opp til 15 % residiv. Pasientene som er behandlet med intracystisk interferon har svært gode resultater hva angår symptomkontroll, men oppfølging blir som ved en kronisk sykdom. Mortaliteten ved kraniofaryngeom er svært lav, men som fremgår er det høy morbiditet.
Oppfølging
Oppfølging avhenger av valgt behandlingsregime. Ved både cystebehandling og tradisjonell kirurgisk behandling er det tett oppfølgning, da residiv forkommer hyppig de første 5 år. Det er risiko for sent residiv, så kontroller (klinisk og MR), bør gjennomføres med lengre intervall gjennom flere tiår.
Nøye symptomatisk oppfølging er helt vesentlig.
Barn/ungdom med kraniofaryngeom kan i varierende grad få senfølger av behandlingen som må følges. Nevropsykologisk testing gjennomføres 1, 2 og 5 år etter diagnose i hht nasjonalt tverrfaglig oppfølgingprofgram for barn med tumor cerebri, og det gjøres en vurdering av behov for dette også tidligere i forløpet. Pedagogisk oppfølging ved behov er vesentlig. Det er en målsetting at der bivirkninger av behandlingen påvises, etableres samarbeid med lokale aktører for å legge til rette best mulig habilitering når det er behov for dette.
Plexus chorioideus-svulster
Epidemiologi
Plexus choroideus svulster utgjør kun 2–4 % av hjernesvulstene hos barn. De fleste pasientene er mellom 0–3 år på diagnosetidspunktet.
Symptomer
Små barn har som oftest en raskt økende hodeomkretsutvikling, eldre barn utvikler generelle trykksymptomer.
Billedundersøkelse
Svulsten er utgående fra plexus choroideus, vevet som produserer CSF i hjernes naturlige hulrom. Svulster i plexus choroideus gir overproduksjon av CSF og ledsagende hydrocephalus er et typisk funn. Hos små barn ser man svulsten i en av sideventriklene eller tredje ventrikkel. Hele CNS-aksen må avbildes for eventuelle metastaser (carcinomer).
Patologi/ Klassifisering
WHO klassifiseringen skiller mellom 3 ulike histologiske grader: Plexus choroideus papillom (WHO grad I), atypisk plexus choroideus papillom (WHO grad II) og plexus choroideus carcinom (WHO grad III).
Diagnosen stilles ved histologisk undersøkelse.
Prognosen
Plexus choroideus papillomer har ved total kirurgisk fjerning en svært god prognose (kurativt).
Plexus choroideus carcinomer har i de fleste rapporter en 5 års overlevelse på under 50 %.
Behandling
Kirurgi er grunnlaget for behandling av plexussvulster hvor målet er total fjerning av tumor. De fleste pasientene er på behandlingstidspunktet i første leveår og har et lite blodvolum, svulstene har stor blodgjennomstrømning, som oftest med en betydelig hydrocefalus. Det kirurgiske inngrepet er derfor utfordrende. Mange vil kreve oppfølgning og eventuell behandling for hydrocephalus i forløpet.
Pasienter med plexus papillom (WHO grad I og II) skal ikke ha tilleggsbehandling, men skal følges opp over tid (regelmessige MR undersøkelser)
Plexus choroideus carcinom (WHO grad III) er en aggressiv tumor. Prognosen er noe bedre ved total fjerning av tumor. Mange har sykdom med spredning og eller innvekst sentralt i hjernen allerede på diagnosetidspunktet og vil være avhengig av tilleggsbehandling.
Plexus choroideus carcinomene er følsomme for kjemoterapi med kombinasjoner av carboplatin, etoposid, vincristin og cyklofosfamid. Stråleterapi forbedrer prognosen, men pasientens alder vil ofte være en begrensende faktor (de fleste vil være for unge). Det er ingen konsensus omkring optimal behandling.
Oppfølging: Alle barn kontrolleres med tanke på tumorkontroll etter 3, 6, 12 18 og 24 mnd med MR, deretter MR minimum to ganger årlig fra 2 til 5 år etter avsluttet behandling, og senere årlig.
Barn med plexuscarcinomer vil i varierende grad utvikle senfølger av behandlingen som må følges nøye. Nevropsykologisk testing gjennomføres i hht nasjonalt tverrfaglig oppfølgingsprogram for barn med hjernesvulst 1, 2 og 5 år etter diagnose, og det gjøres en vurdering av behov for dette også tidligere i forløpet. Pedagogisk oppfølging ved behov er vesentlig.
Det anbefales oppfølging av barnenevrolog, fysioterapeut, endokrinolog, nyrefunksjon, hørsel, ØNH-status og øyestatus. Der bivirkninger av behandlingen påvises, etableres det et samarbeid med lokale aktører for å legge til rette best mulig habilitering.
Solide svulster utenfor sentralnervesystemet
Sist faglig oppdatert: 26.05.2020
Sarkomer
Sarkomer er maligne svulster som utgår fra mesenkymalt vev. Det er den nest vanligste formen for solid, malign svulst utenfor sentralnervesystemet i barnealder (Childhood cancer in the Nordic countries, 2008).
Sarkomer inndeles i bensarkomer og bløtvevsarkomer og får navn etter det vevet de har fellestrekk med.
Bensarkomer inndeles i osteogent sarkom (Kager et al., 2003) og Ewing sarkom, sistnevnte kan også opptre i bløtvev (Principles and practice of pediatric oncology, 2006).
Bløtvevsarkomer inndeles i rabdomyosarkom som utgår fra celler som er eller skal bli muskelceller, og non-rabdomyosarkom. Non-rabdomyosarkom er en heterogen gruppe tumores som består av mer enn 30 ulike undergrupper (Principles and practice of pediatric oncology, 2006).
Se også «Nasjonalt handlingsprogram med retningslinjer for utredning, behandling og oppfølging av sarkom» (Helsedirektoratet, 2018).
Epidemiologi
Insidensen av sarkom hos barn er 1,49 pr 100 000, det vil si at 9,5 % av barna som får en kreftdiagnose, har sarkom. Bløtvevsarkom er hyppigst og forekommer hos 6 % av de kreftsyke barna. Bensarkom er sjeldnere og utgjør 3,5 % av krefttilfellene hos barn under 15 år (Childhood cancer in the Nordic countries, 2008).
Symptomer og funn
Osteogent sarkom debuterer oftest med smerter. I begynnelsen er smertene ofte intermitterende, men etter hvert blir de konstante og mer intense. Tumorrelatert hevelse og nedsatt funksjon i naboledd oppstår mye senere. I ca 10 % av tilfellene debuterer osteogent sarkom som patologisk fraktur (Kager et al., 2003).
Ewing sarkom debuterer oftest med smerter. Smertene er vedvarende og kan oppstå uavhengig av aktivitet, for eksempel i hvile eller om natten. Eventuelle tilleggssymptomer avhenger av tumors lokalisasjon og størrelse. Subfebrilitet er rapportert hos ca 1/3 av pasientene, først og fremst hos de med metastatisk sykdom på diagnosetidspunktet (Paulussen, Kovar, & Jürgens, 2005).
Hvordan et bløtvevsarkom debuterer, avhenger av tumors lokalisasjon og størrelse. Hvis ikke andre strukturer komprimeres, gir sarkom sjelden uttalte symptomer og oppdages ofte tilfeldig grunnet tumors størrelse.
Bløtvevsarkom kan gi smerter hvis de presser på andre strukturer eller obstruerer avløp som for eksempel i blæreregionen. Vedvarende nesetetthet og sekresjon fra nesen er vanlige symptomer ved sarkom i nasofarynks. Vaginale polypper og utflod er vanlig ved sarkom i uterus og vagina (Bisogno & Bergeron, 2005).
Allmenn sykdomsfølelse og vekttap forekommer svært sjelden i startfasen ved sarkom, men forekommer hyppigere ved avansert sykdom.
Utredning
Utredning av sarkom hos barn og ungdom bør skje ved Barne-/ungdomsavdelinger med spesialkompetanse i håndtering av sarkom eller et sarkomsenter. Det er svært viktig at utredningen skjer i henhold til gjeldende protokoller. Man bør mistenke sarkom ved tumor i benvev eller tumor beliggende dypt i bløtvev.
Utredning med tanke på diagnostikk og stadieinndeling gjøres i henhold til aktuelle behandlingsprotokoller og må inneholde adekvat billeddiagnostikk av primærtumor og nøyaktig evaluering av potensielle metastaser.
Radiologiske undersøkelser
Ved mistanke om osteogent sarkom er det viktig med vanlig røntgenundersøkelse av det aktuelle området, gjerne supplert med CT for å se på skjelettstruktur, og MR for å evaluere benmargs- og bløtdelsaffeksjon samt tumors relasjon til årer og nerver. Søk etter metastaser konsentreres om skjelett og lunger for >95 % av metastasene ved osteogent sarkom opptrer i disse lokalisasjonene. Røntgen og CT av lungene benyttes for å kartlegge eventuelle lungemetastaser. MR av affisert knokkel skal utføres for å lete etter skip-metastaser. MR og NaF-PET bør utføres for å kartlegge eventuelle skjelettmetastaser. FDG-PET/CT er en nyttig tilleggsundersøkelse for kartlegging av sykdomsutbredelse ved osteogent sarkom.
Ved utredning av Ewing sarkom benyttes vanlig røntgen, MR og CT for å kartlegge sykdoms-utbredelse på primærtumorstedet. CT egner seg best til å vise endringer i korteks- og skjelettstruktur, mens MR egner seg best for kartlegging av tumorutbredelse i benmarg og bløtvev samt for å evaluere tumors relasjon til nærliggende strukturer som årer og nerver. Det er viktig å dokumentere tumors utbredelse i 3 dimensjoner. CT thorax anbefales for kartlegging av eventuelle lungemetastaser. MR anbefales for kartlegging av eventuelle skjelettmetastaser. FDG-PET/CT er en nyttig tilleggsundersøkelse for kartlegging av sykdomsutbredelse ved Ewing sarkom.
Ved mistanke om bløtvevsarkom bør man benytte MR, ultralyd og eventuelt CT av primærtumor-stedet. Det er viktig å kunne angi tumors størrelse i 3 dimensjoner ved diagnose, slik at man kan evaluere behandlingsrespons ved senere evalueringstidspunkter. MR er å foretrekke for de fleste lokalisasjoner og er obligatorisk ved primærtumor i genitourinaltraktus og ved paraspinal tumor. CT kan være et supplement ved spørsmål om subtile bendestruksjoner. Røntgen thorax og CT thorax benyttes for å oppdage lungemetastaser. Ultralyd abdomen benyttes for å kartlegge lymfeglandelaffeksjon og levermetastaser. FDG-PET/CT er en nyttig tilleggsundersøkelse for kartlegging av sykdomsutbredelse ved bløtvevsarkom.
Biopsi
Sarkomdiagnosen må alltid bekreftes histologisk. Målet med biopsitaking er å få nok tumormateriale til histologi, immunhistokjemi, cytogenetikk, sentralisert «review», samt vev i reserve for eventuelle supplerende analyser.
Biopsitaking skjer oftest ultralydveiledet, men kan også skje CT-veiledet. Det er nyttig å starte med finnålsaspirasjon og fortsette med tru-cut-biopsi hvis forholdene ligger til rette for det. Dersom denne fremgangsmåten ikke kan sikre nok materiale for å kunne stille en sikker diagnose er det aktuelt å supplere med åpen biopsi. Det er viktig å ta med i betraktning at biopsikanalen og eventuelle arr må inkluderes «en-bloc» ved etterfølgende kirurgi, så det er viktig at affeksjon av nevrovaskulære strukturer i størst mulig grad unngås. På ekstremiteter bør incisjoner legges longitudinelt. Farging f.eks. med metylenblått bør benyttes for markering av biopsikanal. Endoskopisk biopsitagning er akseptabelt ved tumor i blære, prostata og vagina.
Benmargsaspirasjon og eventuelt benmargsbiopsi hører med i utredningen av enkelte typer bløtvevsarkom.
Spinalpunksjon for å sjekke om det foreligger CNS-affeksjon, er kun nødvendig ved CNS-nær tumor.
Behandling
Sarkom i barnealder utredes og behandles i henhold til internasjonale protokoller. For øyeblikket behandles osteogent sarkom etter anbefalinger fra den avsluttede EURAMOS 1-protokollen, Ewing sarkom behandles i henhold til EuroEwing 2012-protokollen, mens residiv av Ewing sarkom og refraktært Ewing sarkom behandles i henhold til rEECur-protokollen. Rabdomyosarkom behandles i henhold til anbefalinger fra den avsluttede EpSSG RMS 2005-protokollen og høygradig malignt non-rabdomyosarkom behandles i henhold til anbefalinger fra den avsluttede EpSSG NRSTS 2005 (https://www.skion.nl/). Når det gjelder rabdomyosarkom, har fagmiljøet i Norge søkt REK og SLV om å få godkjent at norske pasienter kan inkluderes i og behandles i henhold til den nye internasjonale behandlingsprotokollen fra EpSSG, nemlig «FaR-RMS: An overarching study for children and adults with Frontline and Relapsed RhabdoMyoSarcoma». Forhåpentligvis kan norske pasienter få tilbud om være med i denne studien allerede i løpet av 2020. Studien er i regi av EpSSG og behandlingen er basert på resultatene fra RMS-2005-protokollen. Behandlingen av sarkom i barnealder er i hovedtrekk multimodal og består av kjemoterapi, kirurgi og/eller bestråling.
Medikamentell behandling
Behandlingen av osteogent sarkom består av pre- og postoperativ kjemoterapi samt kirurgi. Kjemoterapien baserer seg på høye doser metotreksat, cisplatin og doksorubicin (Bielack et al., 2015; Smeland et al., 2019)
Ewing sarkom er generelt en kjemosensitiv tumorform og induksjonsbehandlingen består per i dag av vinkristin, doksorubicin, cyklofosfamid, ifosfamid og etoposid (EE2012-protokollen: https://birmingham.ac.uk/research/activity/mds/trials/crctu/trials/EE2012/index.aspx). Ved dårlig histologisk eller radiologisk respons, ved stor primærtumor (>200 ml) eller ved metastatisk sykdom kan man vurdere å intensivere kjemoterapien med høydosebehandling med autolog stamcellestøtte (https://www.skion.nl/workspace/uploads/Protocol-EpSSG-RMS-2005-1-3-May-2012_1.pdf). Ved behandlingsrefraktær sykdom og residiv består behandlingen av intensiv kjemoterapi og det er flere ulike kjemoterapiformer som har vist seg å ha effekt:
- cyklofosfamid/topotekan,
- irinotekan/temozolamid,
- gemcitabin/docetaxel og
- høydose ifosfamid.
I rEECur-studien randomiseres pasientene mellom de ulike behandlingsarmene for å finne ut hvilken av de fire armene som fungerer best. Foreløpige resultater fra rEECur-studien tyder på at gemcitabin/docetaxel har dårlig effekt og denne behandlingen anbefales derfor ikke.
Rabdomyosarkom er som regel en kjemosensitiv tumorform, og kjemoterapi benyttes derfor i behandlingen hvis man ikke kommer til målet med kirurgi alene. Kjemoterapi benyttes for å redusere tumorstørrelsen slik at radikal kirurgi, evt annen lokalbehandling, kan bli mulig. Aktuell kjemoterapi består av ifosfamid, vinkristin, actinomycin D og eventuelt doxorubicin. Etter avsluttet primærbehandling kan det være aktuelt med vedlikeholdsbehandling med cyklofosfamid og vinorelbin i lave doser (https://www.skion.nl/workspace/uploads/Protocol-EpSSG-RMS-2005-1-3-May-2012_1.pdf). I den nye behandlingsprotokollen (FaR-RMS) vil dessuten irinotekan, temodal og regorafenib inngå i behandlingen av enkelte pasientgrupper.
Non-rabdomyosarkom er en svært heterogen gruppe sarkomer, og de fleste er ikke like kjemo-sensitive som rabdomyosarkom. Den europeiske studien EpSSG NRSTS 2005 (skion.nl/workspace/uploads/EpSSG-NRSTS-version-1-1.pdf) inndeler non-rabdomyosarkom i 3 ulike hovedgrupper; synovialt sarkom, «voksen type» bløtvevsarkom og andre typer bløtvevsarkom. Synovialt sarkom er desidert den vanligste formen for non-rabdomyosarkom i barnealder. Denne sarkomtypen er relativt kjemosensitiv, og hvis det ikke ligger til rette for radikal kirurgi i utgangspunktet, anbefales det kjemoterapi med ifosfamid/doxorubicin for å redusere tumorstørrelsen før eventuell kirurgi. Ved «voksen type» bløtvevsarkom er effekten av kjemoterapi mer usikker, men EpSSG NRSTS 2005 anbefaler kjemoterapi i form av ifosfamid/doxorubicin dersom man ikke kan foreta radikal kirurgi i utgangspunktet (https://skion.nl/workspace/uploads/EpSSG-NRSTS-version-1-1.pdf).
Rabdoid tumor utenfor sentralnervesystemet er en undergruppe av non-rabdomyosarkom. Den er sjelden, men svært aggressiv og ofte letal i barnealder. Grunnet høy dødelighet av denne tumortypen er det svært viktig med adekvat behandling. EpSSG NRSTS 2005 anbefaler et intensivt kjemoterapiregime med vinkristin/doxorubicin/cyklofosfamid alternerende med ifosfamid/etoposid hver annen uke (https://skion.nl/workspace/uploads/EpSSG-NRSTS-version-1-1.pdf). Alternativt kan rabdoid tumor behandles i henhold til anbefalingene fra det multinasjonale European rhabdoid registry (EU-RHAB). EU-RHAB anbefaler et intensivt kjemoterapiregime med doksorubicin, ifosfamid, karboplatin, etoposid, vinkristin, actinomycin D og cyklofosfamid, evt supplert med høydosebehandling med karboplatin og thiotepa, etterfulgt av autolog stamcellestøtte.
Kirurgisk behandling av rabdomyosarkom (RMS)
Lokal kontroll er nødvendig for å kurere barn med RMS, og dette kan oppnås ved kirurgi og/eller strålebehandling (evt brachyterapy). Konservativ tilnærming er anbefalt, og preoperativ kjemoterapi for reduksjon av tumor er vanlig før tumorreseksjon/bestråling.
Målet med lokalbehandling er kurasjon uten eller med minst mulig senskader. Hvilken type lokalbehandling som egner seg best avhenger av primærtumors lokalisasjon og størrelse, pasientens alder og kjemoterapirespons. Kirurgisk planlegging må inkludere alle rekonstruktive prosedyrer med optimal timing av eventuell strålebehandling i tillegg (https://www.skion.nl/workspace/uploads/Protocol-EpSSG-RMS-2005-1-3-May-2012_1.pdf).
Anbefalinger:
- Sarkom i barnealder utredes og behandles i henhold til internasjonale protokoller som krever samtykke fra foreldrene.
- Osteogent sarkom behandles i henhold til anbefalinger i den avsluttede EURAMOS 1-protokollen.
- Ewing sarkom behandles primært i henhold til EuroEwing 2012-protokollen og i henhold til rEECur-protokollen ved residiv.
- Rabdomyosarkom behandles i henhold til anbefalinger i den avsluttede EpSSG RMS 2005- protokollen. I løpet av 2020 vil det være aktuelt med behandling i henhold til EpSSG-studien FaR-RMS.
- Non-rabdomyosarkom behandles i henhold til EpSSG NRSTS 2005-protokollen.
Lymfeknuter
Målet er å påvise lymfeknuteaffeksjon ved «sampling» og unngå radikalt lymfeknutetoilette. «Sentinel node mapping» med for eksempel methylenblått anbefales.
For spesifikke lokalisasjoner og detaljer forøvrig vises det til Protokoll EpSSG RMS2005, versjon 1.4 international – Dec 2013 (https://www.skion.nl/workspace/uploads/Protocol-EpSSG-RMS-2005-1-3-May-2012_1.pdf) og etter hvert også protokollen FaR-RMS
Kirurgisk behandling av non-RMS bløtvevssarkom (non-RMS STS)
Her er det tilnærmet identiske kirurgiske grunnprinsipper som for RMS. Det vises for øvrig til protokoll EpSSG NRSTS 2005 (https://skion.nl/workspace/uploads/EpSSG-NRSTS-version-1-1.pdf).
Kirurgisk behandling av bensarkomer
Prinsippet er at tumor må fjernes med fri margin. Det er viktig med nøye planlegging i forkant av inngrepet. Den kirurgiske behandlingen må passe inn i den multimodale behandlingen uten å forsinke øvrig behandling. Se «Nasjonalt handlingsprogram med retningslinjer for utredning, behandling og oppfølging av sarkom» (Helsedirektoratet, 2018).
Strålebehandling
Dersom man ikke oppnår lokal kontroll med kirurgi, kan det være aktuelt med supplerende strålebehandling. Strålebehandling påvirker vekst og utvikling og skal benyttes med varsomhet til barn og ungdom.
Osteogent sarkom er svært lite strålesensitiv, og strålebehandling inngår derfor ikke i rutinebehandlingen, men benyttes kun ved inoperabilitet og i palliativt øyemed.
Ewing sarkom er svært strålesensitiv. Strålebehandling inngår rutinemessig i behandlingen av Ewing sarkom hvis man ikke oppnår fullstendig lokal kontroll med kjemoterapi og kirurgi, det vil si ved marginal og intralesjonell reseksjon, samt ved inoperabilitet. Strålebehandling benyttes dessuten i behandlingen av lunge- og skjelettmetastaser ved Ewing sarkom.
Rabdomyosarkom er strålesensitiv, og strålebehandling inngår som ledd i behandlingen hvis ikke histologien er lite aggressiv og lokalisasjonen gunstig, eventuelt at kirurgien er komplett. Strålebehandling av rabdomyosarkom gjennomføres fortrinnsvis parallelt med postoperativ kjemoterapi.
I gruppen non-rabdomyosarkom er strålefølsomheten variabel, og det er kun enkelte entiteter som synovialt sarkom, «voksen form» for bløtvevsarkom og rabdoid tumor utenfor sentralnervesystemet som behandles med strålebehandling. Både primærtumor og lymfeglandelmetastaser behandles. Strålebehandling gis fortrinnsvis parallelt med postoperativ kjemoterapi (https://skion.nl/workspace/uploads/EpSSG-NRSTS-version-1-1.pdf).
Oppfølging og etterkontroll
Etter avsluttet behandling for sarkom er det viktig å utføre en sluttevaluering før man starter med regelmessige kontroller. Dette for å evaluere effekten av behandlingen og for å ha et utgangspunkt for senere kontroller. Sluttevalueringen foretas vanligvis 4–6 uker etter avsluttet behandling og omfatter alltid generell klinisk undersøkelse, billeddiagnostikk av affiserte tumorområder, røntgen thorax og blodprøver. Ytterligere detaljer er beskrevet i hver enkelt protokoll. Etterkontrollene skjer vanligvis hver 3. måned de første 2–3 årene, deretter sjeldnere. Hensikten med kontrollene er både å oppdage et eventuelt residiv så tidlig som mulig og å kartlegge eventuelle seneffekter av behandlingen, både på kort og lang sikt. Alle som er behandlet for sarkom, bør følge vanlige kontroller med henblikk på: Høyde og vekt, blodtrykk, Tanner stadium, testikkelvolum, tidspunkt for menarke, skoleprestasjoner, hjertefunksjon, hørsel, nyrefunksjon og eventuelle atferdsproblemer. Detaljer for oppfølging er beskrevet i hver enkelt protokoll.
Se ellers kapittelet om senskader.
Behandling av metastatisk sykdom
Hos enkelte barn er sarkom metastatisk på diagnosetidspunktet. Behandlingen av metastatisk sykdom hos barn baserer seg på de samme prinsipper som ikke-metastatisk sykdom, men kjemoterapien er gjerne enda mer intensiv.
Livsforlengende og palliativ behandling
Ved behov for palliativ behandling hos barn har man ikke lenger et kurativt siktemål, og hensikten med behandlingen vil være å forsøke å lindre plagene så mye som mulig. Dette kan skje ved lavdosekjemoterapi og strålebehandling for å forsinke tumorvekst og lindre smerter, eventuelt smertelindrende medikamenter. Hjemmebehandling og et ordinært liv tilstrebes så lenge som mulig. Viser for øvrig til nasjonale retningslinjer for palliativ behandling hos barn (Helsedirektoratet, 2017).
Nyresvulster
Nefroblastom er den vanligste nyresvulsten i barnealder og kalles til daglig oftest Wilms tumor etter Max Wilms som beskrev den i 1899.
Dette er en ondartet svulst med utgangspunkt i embryonale nyreceller og utgjør ca 90 % av ondartede nyresvulster i barnealder. Opptrer oftest som en enkelt svulst i en nyre, men det kan være multiple svulster, og de kan også opptre i begge nyrer (bilateral svulst). Svulsten diagnostiseres oftest i småbarnsalder og er sjelden hos større barn og ungdommer. Median alder ved diagnose 3,5 år. Over 80 % er under 5 år. Kan også forekomme opp i voksen alder.
Nefroblastom rammer ca 1 av 10 000 barn. Det er ca 6–8 nye tilfeller med nefroblastom i Norge pr år.
Overlevelsen ved nefroblastom er nå gjennomsnittlig ca. 90 % (Pritchard-Jones & Pritchard, 2004).
Genetikk
Nefroblastom forkommer enten sporadisk, i assosiasjon med visse syndromer (se senere) og hos 1–2 % av pasientene som familiær sykdom. Ved familiær sykdom synes genetikken å være kompleks med flere mulige gener involvert.
Kartlegging av genetikk og molekylærbiologi ved nefroblastom er i stadig utvikling. Og man venter at nyere molekylærbiologisk forskning vil kunne identifisere genetiske endringer av betydning for risikoinndeling, behandling og prognose både ved sporadiske og arvelige former. Det vises også til kapittelet Genetikk.
Nefroblastomatose: Betegner en tilstand hvor det i den tidlige nyreutviklingen har blitt værende igjen umodne små foci av embryonale celler (nefrogenic rests) i nyreparenchymet (Beckwith, 1998). Disse vil kunne utvikle seg videre til større hyperplastiske knuter og evt til nefroblastomsvulster. Disse oppdages enten ved at de er så store (hyperplastiske) at de sees som knuter i nyrene på ultralyd ved utredning av nyretumor, eller som tilfeldig funn. De kan også være helt mikroskopiske og finnes ved undersøkelse av gjenværende nyrevev i nyre fjernet grunnet tumor. Det vil da være økt risiko for ny tumor i gjenværende, kontralaterale nyre.
Diagnose
I nesten 50 % av tilfellene har barna ingen symptomer, og svulsten oppdages da tilfeldig enten i forbindelse med andre undersøkelser i helsetjenesten eller ved at foreldre eller andre kjenner at det er en kul i barnets mage. Vel 50 % vil ha magesmerter, få stor, utspilt mage eller ha blod i urinen. Og ca 5 % vil ha tydelig nedsatt allmenntilstand.
Høyt blodtrykk, trombocytose eller hevelse i pungen kan være ledsagende symptomer.
Diagnostikken består av full klinisk undersøkelse hvor man spesielt må være forsiktig ved palpasjon av abdomen, og unngå å trykke hardt på svulsten som kan være skjør. Det er ingen spesifikk tumormarkør knyttet til nefroblastom. Diagnosen stilles ved bildediagnostikk, ultralyd og MR. Fordi 90 % av alle ondartede svulster i nyre er Wilms tumor er det vanlig å starte behandling med cellegift uten forutgående vevsprøve. Vevsprøve forbeholdes de tilfeller hvor diagnosen er usikker etter MR, hvor barnet er yngre enn 7 måneder eller eldre enn 16 år.
Tilstander som disponerer for nefroblastom
Nefroblastom er sterkt assosiert med enkelte syndromer/utviklingsforstyrrelser som:
- Aniridi
- Hemihypertrofi
- Genital/urinveis-midannelser
- Beckwith-Wiedemanns syndrom
- Denys-Drash syndrom
- WAGR syndrom
Pasienter med disse syndromene/utviklingsforstyrrelsene skal kartlegges og tilbys jevnlig kontroll der dette er indisert.
Klassifisering
Klassifiseringen følger den behandlingsprotokollen vi nå bruker:
Umbrella-protokoll SIOP-RTSG 2016. Studien vil åpnes i Norge i løpet av kort tid, men allerede nå brukes den som grunnlag for klassifisering og behandling. Denne protokollen har retningslinjer for behandling av alle typer nyresvulster.
Det er nasjonal enighet om å delta i dette multinasjonale samarbeid for nyresvulster hos barn i regi av SIOP. Det er etablert en organisasjon SIOP Renal Tumour Study Group (www.siop-rtsg.eu) som leder arbeidet. Norge deltar der sammen med de fleste nordiske land. SIOP har gjennomført kliniske studier for nefroblastom fra 1971. Det har vært økende tilslutning av deltagende land. Norge deltok tidligere mest i de engelske nefroblatomstudiene, og sluttet seg, på nasjonal basis, til SIOP-studiene da England også gjorde det fra 2001.
SIOP Renal tumour Study Group har også et nært samarbeid med nyretumorgruppen i COG (Children’s Oncology Group) i USA (http://www.childrensoncologygroup.org/, www.curesearch.org)
Den største forskjell i behandling mellom studiene til SIOP-RTSG og COG er at SIOP-RTSG foretrekker preoperativ kjemoterapi for om mulig å skrumpe tumor før kirurgi og gjøre den lettere håndterbar, mens COG går direkte til primær kirurgi der dette er mulig. De medikamentene som brukes er vinkristin, aktinomycin D og for enkelte grupper også doxorubicin (Kaste et al., 2008; Pritchard-Jones & Pritchard, 2004).
Vår klassifisering bygger på utbredelse etter preoperativ kjemoterapi, og endelig stadiesetting kommer først etter den utsatte kirurgien.
Ved diagnosetidspunktet deles pasientene inn i følgende kategorier:
- lokalisert sykdom
- metastatisk sykdom
- synkron bilateral sykdom
Preoperativ behandling gis ut fra dette. Diagnosen vil oftest baseres på bildediagnostikk alene. Biopsi/cytologi kan bli nødvendig ved usikre funn på MR eller dersom barnet er yngre enn 7 måneder eller eldre enn 16 år, men det er i henhold til den nyeste behandlingsprotokollen altså unødvendig i de fleste tilfeller.
Ultralyd av abdomen er obligatorisk. Metoden kan skille ut cystiske komponeneter og er nyttig for å oppdage små tumores i kontralateral nyre, tumorinnvekst i nyrevene og vena cava, samt metastaser til lever og abdomen. Man får også mål til volumberegning av svulsten ved diagnose. Samme mål gjentas preoperativt etter kjemoterapi.
Rtg.thorax front og side samt CT thorax er også obligatorisk i siste behandlingsprotokoll.
Videre suppleres bildediagnostikken med MR abdomen (alternativt CT abdomen) og evt preoperativ MR eller CT angiografi (for framstilling av blodkar og deres relasjon til tumor).
Nålebiopsi ved nyretumor hos barn skal kun gjøres ved usikkerhet rundt diagnosen etter radiologisk utredning, ved alder yngre enn 7 måneder eller eldre enn 16 år.
Biopsi med Tru-Cut nål utføres av radiolog i generell anestesi, og med patolog til stede for å sikre adekvat vevsmengde. Sykehusets retningslinjer for intervensjon må følges.
Ved bilateral sykdom eller mistanke om nefroblastomatose er det egne anbefalinger med ytterligere utsettelse av kirurgien ved forlenget preoperativ behandling – se protokoll.
Postoperativ behandling: Hele den postoperative behandlingsstratifiseringen hviler på patologens stadieinndeling og histologiske vurdering av operasjonspreparatet etter preoperativ behandling.
| Stadium 1 | Lokalisert til nyren, radikalt fjernet |
| Stadium 2 | Tumorvekst utenfor nyren, men radikalt fjernet |
| Stadium 3 | Tumorvekst utenfor nyren, ikke radikalt fjernet |
| Stadium 4 | Fjernmetastaser (oftest i lungene) |
| Stadium 5 | Tumor i begge nyrer (Bilateral sykdom). |
Histologien deles i lav-, intermediær- og høyrisiko etter bestemte kriterier.
Kombinasjonen av stadium og histologisk risikogruppe bestemmer så hvilken postoperativ kjemoterapi som skal gis, og hvorvidt det også skal suppleres med strålebehandling – se protokoll for detaljer.
Umbrella-protokoll SIOP-RTSG 2016 har som mål gjennom internasjonalt samarbeid å samle pålitelige data som kan brukes til å besvare spørsmål som har direkte klinisk relevans for pasienter. Man ønsker å samle tumorvev samt blod og urin for å lete etter nye markører for å forbedre en risikotilpasset stratifisering av behandlingsintensiteten. Protokollen vil åpnes for å inkludere norske pasienter innenfor kort tid.
Prognosen ved nefroblastom nå er så god at man vil også forsøke å designe behandlingsprogrammer som reduserer risiko for senskader (antracykliner innebærer en risiko for kardiale senskader), og opprettholder de gode behandlingsresultatene.
Andre målsettinger i studien er å forsøke å bedre resultatene ved å optimalisere seleksjonen (riktig stadie/riktig histologi) ved sentral «review» av både histologi og radiologi på alle pasienter
Langtidsoppfølging av barn med nyresvulster
Kirurgi, kjemoterapi og eventuell strålebehandling kan gi senfølger som manifesterer seg kort eller lang tid etter behandling. Nefroblastompasienter har økt risiko for hypertensjon og nedsatt nyrefunksjon. Strålebehandling vil gi senfølger av ulik grad avhengig av omfanget for strålebehandlingen (påvirkning av tarmfunksjon, skoliose, lungeforandringer ved stålebehandling av metastaser, sekundær cancer m.m). Bruk av antracykliner i behandlingen samt evt bestråling av thorax er risikofaktorer for kardiale senskader (Kfr Medisinsk støttebehandling kap. 10) (Wright, Green, & Daw, 2009).
Tilbakefall av nefroblastom
I den nye Umbrella 2016-protokollen har RTSG sammen med COG utarbeidet retningslinjer for behandling av tilbakefall av nefroblastom. Tilbakefallene er sjeldne i en sykdom med så god prognose. De representerer likevel store utfordringer, og økt behandlingsintensitet medfører også risiko for langtidseffekter av behandlingen.
Her brukes som basis de samme medikamentene, vinkristin og actinomycin D (Spreafico et al., 2009) som i primærbehandling med tillegg av andre medikamenter som cyklofosfamid, etoposid og carboplatin. I noen tilfeller anbefales høydosebehandling med stamcellestøtte. I tillegg må mange ha strålebehandling.
Behandling av nefroblastomatosepasienter
Disse pasientene er i økt risiko for å utvikle nefroblastom og skal ha spesielt oppfølgingsprogram og behandling. Det gis ofte langvarig kjemoterapi (minst ett års varighet) inntil forandringene forsvinner eller inntil det blir mulig å fjerne dem med kirurgi. Etter en slik operasjon må det også gis langvarig kjemoterapi.
Andre nyresvulster
I tilegg til en rekke benigne svulster kan det oppstå andre maligne svulster i nyrene; Klarcellet nyresarkom (CCSK), «malign rhabdoid tumour of the kidney» (MRTK), nyrecarcinom samt metastaser fra annen kreftsykdom.
Øvrige retningslinjer som finnes i protokollen
- Behandling for pasienter med primær kirurgi.
- Behandling av bilateralt nefroblasom (gjelder ca 5 % av pasientene).
- Opplegg for behandling av nefroblastomatose.
- Behandling av clear cell sarcoma of the kidney, rhabdoid tumour of the kidney og residiverende sykdom.
- Retningslinjer for oppfølging.
- Behandling av nefroblastom hos voksne.
Kirurgisk behandling av nefroblastom
Det er enighet i det norske barneonkologiske miljøet om nasjonal deltakelse i Umbrella-protokoll SIOP-RTSG 2016, og studien er godkjent av etisk komite. Studien er fortsatt ikke åpnet for inklusjon av norske pasienter, men som «best available treatment» behandler vi i henhold til denne selv om pasientene ikke kan inkuderes i studien. Behandlingen er altså risiko-adaptert og det er viktig å følge protokollen i dens retningslinjer både for kirurgien, stadiesettingen og rapporteringen (sistnevnte kun når pasienter inkluderes). Behandlingsteamet må kjenne behandlingsprotokollen og ha meldeskjemaene for kirurgi for hånden før og/eller under operasjonen.
Tumor i én nyre
Ved operasjon for Wilms’ tumor prioriteres god tilgang fremfor kosmetisk, begrenset snitt. Det anbefales bred, transversal incisjon ovenfor umbilicus og tilgang via laparatomi. Minimal invasiv kirurgi (kikkhull/laparoskopi) er så langt ikke aktuelt hos barn. Bukhulen inspiseres og palperes nøye med tanke på spredning. Moderne billedfremstilling preoperativt gjør det unødvendig å åpne peritoneum/nyrefascien over motsatt sides nyre hvis det ikke er påvist suspekte forandringer i den.
Et generelt prinsipp ved tumorkirurgi er å benytte såkalt «non touch / minimal touch»-teknikk før drenerende kar fra tumor er avstengt. Om mulig bør arterien avklemmes først. Binyren fjernes sammen med nyren hvis ikke tumor er i god avstand fra den. Lymfeknute-sampling er viktig, kfr. behandlingsprotokollen, men radikalt lymfeknutetoilette er (for tiden) ikke indisert. Hvis det foreligger gjennomvekst av kapsel, må en sørge for god margin ut i normalt vev. Heroiske og mutilerende inngrep anbefales imidlertid ikke siden de fleste nyresvulster er både kjemo- og strålesensitive. Preoperativ cytostaticabehandling reduserer sjansene for kapselruptur ved uttak av tumor, men kan vanskeliggjøre bedømmelsen av tumorgrense p.g.a. nekrose/inflammasjon. NB! Sjekk tumors utbredelse på bilder tatt før behandling!
Tumortrombe i vena renalis eller vena cava skal fjernes via venotomi; om tromben når til atriet kan cardiopulmonal bypass være nødvendig.
Nefronsparende tumorreseksjon i stedet for radikal nefrektomi anbefales foreløpig ikke ved ensidig tumor.
Bilateral nyretumor
Ved bilateral tumor bør «nefronsparende» operasjon tilstrebes. Helt avhengig av tumors utbredelse står valget mellom lokal tumorexcisjon bilateralt eller nefrektomi på den ene siden og begrenset reseksjon på den minst affiserte siden. Enukleasjon av små tumores kan være aktuelt, spesielt hvis det er multiple slike i en nyre.
Fjernmetastaser
Lokale levermetastaser skal excideres i forbindelse med primæroperasjonen. Ved utbredte metastaser i leveren skal det tas biopsi. Lungemetastaser skal excideres, men inngrepet kan med fordel utsettes et par uker til resultatene av primæroperasjonen foreligger. Følg for øvrig gjeldende behandlingsprotokoll.
Nevroblastom
Nevroblastom er en ondartet svulst med utgangspunkt i embryonale perifere nerveceller som vanligvis diagnostiseres hos små barn, men sporadiske tilfeller kan også forekomme hos ungdom og unge voksne. I Norge diagnostiseres 6–10 nye tilfeller årlig, og rundt halvparten opptrer som metastatisk sykdom. De fleste nevroblastomer har utgangspunkt i binyrene, men svulsten kan oppstå alle steder der det er sympatisk nervevev (dvs grensestreng og ganglier). Prognosen ved nevroblastom varierer fra nær 100 % overlevelse ved lokal sykdom og gode prognostiske faktorer til rundt 50 % overlevelse hos barn over 1 år med metastatisk sykdom. Dårligst prognose finner vi hos barn > 18 mnd på diagnostidspunktet. Av den grunn er det stor variasjon i behandlingsintensitet hos barn med nevroblastom, fra kun observasjon i de enkleste tilfellene til multimodal intensiv terapi med cellegift, kirurgi, høydosebehandling med stamcellestøtte, stråling og immunterapi hos barn over 1 år med metastatisk sykdom. I Norge behandles nærmest alle barn med nevroblastom etter SIOPEN-protokoller. I SIOPEN samarbeider en rekke land for å oppnå de beste vitenskapsbaserte behandlings- og forskningsprotokoller. Norge er et aktivt medlem av SIOPEN, sammen med 24 andre medlemsland (https://www.siopen.net/).
Diagnose
Symptomer ved nevroblastom er svært varierte og inkluderer skjelettsmerter, fraktur, økt bukomfang, høyt blodtrykk, svette, diaré, nevrologiske utfall, periorbital blødning, Horner syndrom og påvirkning av perifere hematologiske verdier. Diagnosen stilles ved biopsi/cytologi av tumor eller benmargsmetastaser. Forhøyede urinkatekolamin-metabolitter (HVA og VMA), NSE og positive funn ved den nukleærmedisinske undersøkelsen mIBG styrker mistanken om nevroblastom før den cytologiske/histologiske diagnosen foreligger. PET-CT kan gi supplerende informasjon, men anses ikke som nødvendig ved positiv mIBG scan. Det er viktig å sikre nok tumorvev for biologiske analyser (f.eks MYCN-amplifikasjon, kromosomale delesjoner og gains i tumorvev) som kan predikere prognose og nødvendig behandlingsintensitet.
Radiologisk utredning
Siden klassifiseringen baserer seg på den radiologiske utredning med MR og ultralyd (Monclair et al., 2009), er det svært viktig at de preoperative undersøkelsene utføres ved det behandlende sykehus, etter faste protokoller, slik at sammenlikningen av undersøkelsene blir enklere.
Klassifisering
Tidligere stadieinndeling 1–4(S) er med nye protokoller blitt erstattet av den internasjonale INRG-klassifiseringen som forholder seg til risikofaktorer for kirurgi, metastaser og alder (Cohn et al., 2009; Monclair et al., 2009; Pinto et al., 2015);
| L1: | operabel tumor bedømt ut fra radiologiske risikofaktorer |
| L2 | inoperabel tumor bedømt ut fra radiologiske risikofaktorer |
| M: | metastaser til stede |
| MS: | barn under 1 år (18 mnd) med metastaser kun lokalisert til hud, lever og/eller benmarg |
Ung alder og gunstig histologi er forbundet med god prognose, mens biologiske faktorer som segmentale kromosomale aberrasjoner i tumorceller har størst betydning for utfallet av kreftsykdommen. I denne sammenheng er N-Myc amplifikasjon, 11-q delesjon, 1-p delesjon og 17-q gain de viktigste kromosomale avvik. N-Myc amplifikasjon er vanligst hos de minste barna, mens 11-q delesjon har størst betydning for utfallet ved nevroblastom hos eldre barn. Denne erkjennelsen har medført at biologiske faktorer tas hensyn til ved klassifisering og planlegging av behandlingen ved nevroblastomsykdom hos barn.
Behandling
Intensiteten i behandlingen av nevroblastom varierer avhengig av prognostiske faktorer (Pinto et al., 2015). Ytterpunktene demonstreres ved at man ved stadium MS uten livstruende symptomer eller MYCN-amplifikasjon kun observerer, mens man ved stadium M tar i bruk all tilgjengelig målrettet medisinsk behandling (kjemoterapi, kirurgi, høydosebehandling med stamcellestøtte, stråling, vitamin A og immunterapi med spesifikke GD-2 antistoffer). Kjemoterapien består av cisplatin, karboplatin, cyklophosfamid og vinkristin i induksjonsfasen, evt supplert med topotecan, doksorubicin, temozolomid eller irinotecan dersom induksjonsbehandlingen ikke har tilstrekkelig effekt. Nevroblastom har blitt oppfattet som en kreftform der kirurgien er avgjørende. Det er fortsatt tilfelle, men oppdagelsen av biologiske risikofaktorer har medført at enkelte «gunstige» svulster kan observeres uten kirurgi i første omgang. Følgende SIOPEN-protokoller brukes ved de ulike stadier:
| L1: | ingen åpen studie protokoll, kun kirurgisk behandling |
| L2: | protokoll SIOPEN LINES |
| M: | HR-NBL-1 SIOPEN |
| MS: | protokoll SIOPEN LINES / HR-NBL-1 SIOPEN ved MYCN-amplifikasjon |
Internasjonalt er bruken av immunterapi med GD-2 antistoff økende med lovende resultater (Yu et al., 2010). Dinutuximab Beta ble godkjent i Beslutningsforum juni 2019 til behandling av høyrisiko nevroblastom og relapserende/refraktær nevroblastom.
Ved høyrisk nevroblastom kombineres all tilgjengelig effektiv terapi i første behandlingsrunde. Av den grunn er det per i dag lite dokumentert behandling tilgjengelig i residivsituasjoner. Forskningen er intens på dette fagfeltet, og man har håp om at nye behandlingsformer blir dokumentert effektive i nær framtid. Høydose MIBG-behandling med eller uten topotekan-sensitivisering (Gaze et al., 2005) har vist seg effektiv i enkelte situasjoner og kan nå tilbys i Norge i en residiv- eller refraktær situasjon. For høyrisiko nevroblastomer i en residiv/refraktær situasjon der det foreligger informasjon om en ALK abberasjon i tumor, kan det være indisert med behandlingsforsøk med ALK-hemmer (fortrinnsvis en første generasjons ALK hemmer, men noen spesifikke ALK abberasjoner har kjent resistens mot første generasjons ALK hemmere). Denne behandlingen bør fortrinnsvis kombineres med annen tumorrettet behandling. ALK hemmer inngår per 2020 i førstelinjebehandling av høyrisk nevroblastom med ALK abberasjon i USA (Childrens Oncology Group protokoll ANBL 1531). I Europa har man i første omgang ikke inkludert dette i primærbehandlingen, men det brukes i utstrakt grad i en residivsituasjon.
Kirurgi ved nevroblastom
Kirurgisk behandling av nevroblastom vil som regel være radikal fjernelse av tumor. Dette bør skje uten mutilering, skade på vitale organer eller alvorlig annen morbiditet. For svulster med gunstige biologiske markører kan det være akseptabelt å la en liten resttumor bli værende igjen.
Kirurgi er lokalbehandling. Kirurgiens plass i behandlingen må derfor vurderes ut fra om det kan oppnås radikal fjernelse eller ikke. Det er viktig med god kartlegging av tumors utbredelse, både ved diagnosetidspunkt og preoperativt.
Selv om mange nevroblastomsvulster er svært følsomme for kjemoterapi, vil man med unntak av enkelte spedbarn, ikke oppnå at primærtumor forsvinner helt.
Preoperativ kjemoterapi vil ofte være aktuelt, både for å sanere metastaser og for å øke muligheten for radikal fjernelse av tumor. Ved tilstedeværelse av risikofaktorer bør det gis preoperativ kjemoterapi. Ved metastatisk sykdom vil det også gis preoperativ kjemoterapi selv om primærtumor er uten risikofaktorer, med mindre radikal fjernelse av primærtumor anses å være det beste alternativet for vevsdiagnostikk.
Preoperativ kjemoterapi gjør svulstene mindre, mer fibrøse, mindre vaskulariserte og generelt lettere å håndtere. Dette reduserer risiko for spredning peroperativt, og det er akseptabelt å dele tumor under operasjonen og ta den ut bitvis. Det er ofte nødvendig å dele tumor peroperativt fordi tumor ofte omslutter kar som må bevares. Svulstene kan være kraftig adherente til karveggen, men infiltrasjon i selve karveggen er svært sjelden. Etter kjemoterapi er restsvulsten ofte dårlig definert, og det er umulig peroperativt å skille mellom aktivt nevroblastomvev og modent ganglionevrom, fibrose eller arrvev. Derfor vil man ved operasjonen ha som målsetning å fjerne vev i primærtumors utbredelsesområde, men unngå å skade vitale strukturer. Det vanligste er å operere med åpen kirurgi, men for enkelte lokaliserte svulster kan det være aktuelt med laparoskopi eller torakoskopi.
Komplikasjoner relatert til nevroblastomoperasjoner avhenger av utbredelse og lokalisasjon. Ved inngrep på hals og øvre thorax kan det oppstå nerveskader (eks Horners syndrom). I nedre thorax er det en viss risiko for skade på medullas blodforsyning. Retroperitoneale operasjoner kan gi postoperativ diare og ascites. Ved ekstirpasjon av sentrale abdominale nevroblastomer er det risiko for nyreischemi. Nevroblastomkirurgi i bekkenet har risiko for nerveskade, både til blære, tarm og underekstremiteter.
Toksisitet/seneffekter etter nevroblastombehandling
Metastatisk høyrisk nevroblastom krever intensiv behandling, og dermed er risikoen høy for akutt toksisitet og alvorlige seneffekter både forårsaket av grunnsykdommen og behandlingen. Nevroblastomets lokalisering øker ytterligere risikoen for varige seneffekter, siden svulsten ofte infiltrerer nyre eller vokser inn i spinalkanalen. Likevel er det behandlingsinduserte seneffekter av hørsel og nyrefunksjon som er de vanligste følgetilstandene hos langtidsoverlevere etter intensivt behandlet nevroblastom.
Oppfølging/kontroller
Pasientene følges i minst 10 år etter endt behandling. Tumorovervåking baserer seg på ultralyd av tumorlokalisasjon (evt. MR dersom området ikke er tilgjengelig med ultralyd) og urinkatekolaminer. Radiologisk oppfølging av primærtumor kan avsluttes fem år etter endt behandling. Hørsel og nyrefunksjon er av de viktigste seneffektene å monitorere minst 1, 5 og 10 år etter gjennomført behandling.
Leversvulster
2/3 av leversvulstene i barnealder er ondartede. Dette er sjeldne tilstander og utgjør ca 1–2 % av barnekrefttilfellene. Totalt ser vi 0–3 nye tilfeller i Norge pr år.
90 % av de ondartede svulstene i lever er enten hepatoblastom (HB) eller hepatocellulært carcinom (HCC). HB forekommer langt hyppigere enn HCC (Ratio 1,8: 1)
Fordi svulstene er så sjeldne, er det på verdensbasis dannet samarbeidsgrupper som sammen utvikler programmer for diagnostisering og behandling samt forskning på disse svulstene.
I Norge er vi tilsluttet SIOPs levertumorgruppe SIOPEL (https://siope.eu/news/siopel-patientsfamilies-forum-liver-tumours-children/) som har ledet multinasjonale, kliniske studier på leversvulster hos barn siden 1990. Mer enn 30 land deltar i dette SIOPEL–samarbeidet.
Andre store grupper for behandling av leversvulster hos barn er:
- COG (Children’s Oncology Group), USA (www.childrensoncologygroup.org and www.curesearch.org)
- Levertumorgruppen i GPOH (German Society of Pediatric Hematology and Oncology), Tyskland/Østerrike.
- JCCG – The Japanese children’s Cancer group, Japan
Felles for HB og HCC er at de kan produsere og skille ut alfa-fetoprotein (AFP), et glykoprotein som syntetiseres i plommesekk og lever fra tidlig i fosterlivet. Produksjonen stopper normalt ved fødsel, og konsentrasjonen synker fra gjennomsnittlig 50 000 ng/mL ved fødsel til < 20 ng/mL ved 6–8 måneders alder. Variasjonsbredden i AFP-nivå er stor, og det anbefales at man slår opp i tabell i aktuell protokoll. Helt nede på voksne verdier er man først ved 1–2 års alder.
AFP er forhøyet hos over 90 % ved HB og hos vel 50 % ved HCC.
Hepatoblastom
HB er en ondartet svulst med utgangspunkt i embryonale leverceller. Median alder ved diagnose er 1 år. 80 % sees i de 3 første leveår. HB er også beskrevet opp i ungdoms- og voksenalder. HB opptrer oftest som en solitær leversvulst, men kan være multifokal og da affisere store deler av leveren.
Genetikk. HB opptrer oftest sporadisk, og der vet man foreløpig lite om genetiske og molekylærbiologiske faktorers betydning for utvikling, risikoinndelig, behandling og prognose. HB ses også i assosiasjon med visse syndromer hvor noe mer av genetikken er kjent.
Tilstander som disponerer for hepatoblastom
- Isolert hemihypertrofi
- Beckwith-Wiedemann syndrom
- Familiær adenomatøs polypose
Pasienter med disse syndromene/utviklingsforstyrrelsene skal kartlegges og tilbys jevnlig kontroll. Man skal også se nøye etter tegn på disse tilstandene ved diagnostisert HB.
Økt risiko for HB ses også som en assosiasjon til prematuritet med svært lav fødselsvekt.
Diagnose
HB presenterer seg oftest som en asymptomatisk, stor oppfylning i magen hos et ellers friskt barn. Noen har også allmennsymptomer i form av feber, vekttap, anorexi og anemi. Hos noen sees det også koagulasjonsforstyrrelser med blødning ved diagnose.
Laboratorieutredning viser ofte trombocytose og oftest forhøyet AFP-verdi. I sjeldne tilfeller kan også en annen tumormarkør, beta-chorionic gonadotropine (β-HCG) være forhøyet med økt risiko for tidlig pubertetsutvikling.
Utredning
- Grundig anamnese og generell klinisk undersøkelse
- Blodprøver med tumormarkørpanel (AFP, b-HCG, LD, koagulasjonsprøver, evt andre)
- Ultralyd abdomen
- MR eller CT av abdomen
- Rtg thorax og CT thorax
- Evt MR eller CT caput
- Urin til katekolaminbestemmelse (differensialdiagnostikk)
Diagnostisk biopsi
Biopsi gjøres nå som regel som ultralydveiledet tru-cut biopsi som utføres av radiolog i generell anestesi, og med patolog til stede for å sikre adekvat vevsmengde. Det kan også være nødvendig med kirurgisk biopsi hos noen, avhengig av tumors beliggenhet i leveren.
Biopsi er sterkt anbefalt for alle pasienter med mistenkt malign levertumor med mindre det foreligger blødningsforstyrrelser eller kritisk organpåvirkning.
Histologi
Hepatoblastomene deles inn i epiteliale undergrupper: Fetal, embryonal, makrotrabekulær og småcellet udifferensiert (tidligere kalt anaplastisk) og i blandet epitelial og mesenkymal morfologi med eller uten teratoide trekk.
Klassifisering og stadieinndeling
Det har tidligere blitt benyttet flere ulike systemer for klassifisering og stadieinndeling internasjonalt. I USA og flere andre land har det vært vanlig å gjøre primær kirurgi der dette er mulig, og deres inndeling og stadiesetting har vært basert på dette. Dette er forskjellig fra den europeiske tilnærmingen til behandling, noe som har gjort det vanskelig å sammenfatte og sammenligne forskning på dette feltet.
Den pågående studien på leversvulster som Norge også deltar i er imidlertid et samarbeidsprosjekt mellom den europeiske gruppen (SIOPEL), den amerikanske gruppen (COG) og den japanske gruppen (JCCG) kalt PHITT (paediatric hepatic international tumour trial), og man vil i denne studien undersøke kvaliteten på et felles klassifiseringssystem og behandling. Hovedmedikamentet som har vist seg svært effektivt på leverkreft er cisplatin. Dette gis ofte i kombinasjon med doxorubicin og noen ganger også alternerende med carboplatin. Klassifiseringen følger et skjema utviklet av SIOPEL hvor svulstene blir inndelt etter «PRE-Treatment EXTent of disease» (forkortet PRETEXT) Systemet baserer seg på radiologisk påvist affeksjon av leveren ved diagnose i forhold til leverens anatomi med inndeling i 4 seksjoner (8 leversegmenter) samt innvekst i kar og evt tilstedeværelse av metastaser(For beskrivelse av leveranatomi vises det til SIOPEL publikasjoner og protokoller) (Aronson et al., 2005).
Svulstene deles i:
PRETEXT I: kun 1 seksjon involvert
PRETEXT II: 2 «naboseksjoner» involvert
PRETEXT III: 2 seksjoner som ikke ligger inntil hverandre eller 3 «naboseksjoner» involvert
PRETEXT IV: alle 4 seksjoner involvert
Videre inndeles pasientene i very low, low, intermediate og high risk ut fra følgende faktorer:
Tumoregenskaper (ruptur, fokalitet, innvekst i blodkar, vekst utenfor lever, ruptur/blødning)
Metastaser
Barnets alder
AFP-verdi
Tumors resektabilitet
Maligne leversvulster metastaserer først og fremst til lunger og lymfeknuter, men det er også sett spredning til skjelett og CNS. Leversvulstene metastaserer ikke til benmarg.
Behandling
HB er fortsatt en kirurgisk sykdom. Noen få pasienter er primært operable, men strategien for de fleste risikogruppene er preoperativ kjemoterapi med utsatt kirurgi.
Mange pasienter kan få tumor fjernet med leverreseksjon etter preoperativ kjemoterapi. I tillegg kan enkelte, tidligere inoperable pasienter tilbys levertransplantasjon. Kjemoterapien totalt er med på å hindre tilbakefall for de aller fleste. Til sammen har dette gjennom 20 år gitt en betydelig bedret prognose (Perilongo et al., 2009; Perilongo et al., 2004; Pritchard et al., 2000). Se også kapittelet om Kirurgi.
I Norge brukes PHITT-protokollen som klassifiserings- og behandlingsretningslinje, enten barnet deltar i studien eller ikke.
Overlevelse (overall survival) varierer med risikogruppe, og er for standard risiko HB 95 % og for høyrisikogruppen ca 80 % (Zsiros et al., 2013).
Hepatocellulært carcinom
HCC sees oftest hos litt eldre barn (median alder ved diagnose 12 år) og er en langt alvorligere tilstand. HCC forekommer hyppigere i områder hvor hepatitt B virus (HBV) er endemisk, og da oftest hos barn over 5 år. HCC er også assosiert med hepatitt C virus (HCV)
Tilstander som disponerer for HCC
Underliggende leversykdommer som bl.a. tyrosinemi, biliær atresi, idiopatisk neonatal hepatitt, a1‑antitrypsinmangel, nevrofibromatose, ataxia teangiectasia er assosiert med HCC.
Diagnose
Klinisk bilde som ved HB, men positiv AFP kun hos ca 50 %.
Utredning
Som ved HB. I tillegg utvidede tester med fokus på underliggende leversykdom.
Diagnostisk biopsi
Biopsi (ultralydveiledet) bør gjøres hos alle med mindre tumor kvalifiserer for primær kirurgi.
Histologi
Hepatocellulære carcinomer deles histologisk i pediatrisk, adult og fibrolammelær HCC.
Klassifisering og stadieinndeling
Inndeling i PRETEXT-grupper som ved HB – se over.
Maligne leversvulster metastaserer først og fremst til lunger og lymfeknuter, men det er også sett spredning til skjelett og CNS. Leversvulstene metastaserer ikke til benmarg
Behandling
Behandling av HCC er en utfordring, og suksess i behandlingen er totalt avhengig av mulighetene for kirurgi. Dersom tumor er operabel, skal pasienten ha primær kirurgi da HCC er en relativt kjemoresistent tumor. Under halvparten er operable ved diagnose. Pasienter som ikke er operable ved diagnose eller som har mikroskopisk restsykdom etter diagnose vil randomiseres til å motta en av to ulike kjemoterapiregimer. Målet med dette er å finne en behandling som gir reduksjon i tumor slik at flere blir operable, enten ved reseksjon eller ved transplantasjon, og at flere pasienter blir kurert. Tilbakefall av sykdom ved HCC er også en stor utfordring til dags dato.
Generelle kriterier for levertransplantasjon har til nå vært strenge. Hos voksne pasienter aksepteres ikke svulster med diameter > 5 cm eller multifokalitet med mer enn 3 svulster, spesielt ikke hvis en av dem er > 3 cm eller kombinert diameter er > 8 cm (Milan criteria), men man gjør oftest en mer individualisert vurdering for barn. Dog er begrenset sykdom fortsatt et kriterium for suksess også ved transplantasjon. Metastatisk sykdom vil være kontraindikasjon for transplantasjon.
5 års overall survival for HCC i tidligere SIOPEL-protokoller har vært under 30 %. Man har til nå brukt de samme medikamenter ved HCC som ved hepatoblastom, men uten like stor suksess. Man er nå inne i en fase med mer eksperimentell medikamentell behandling i håp om et gjennombrudd for disse pasientene. I PHITT-protokollen sammenlignes behandling med Gemcitabin og Oxaliplatin med konvensjonell HB-behandling. I tillegg er det lagt til Sorafenib i behandlingen for alle pasientene. Dette er et medikament som har vist effektivitet hos voksne pasienter med HCC. I senere studier kan aktuelle medikamenter være bevacizumab, everolimus, sirolimus osv. Lokal intervensjons-behandling direkte mot tumor med radioembolisering eller medikamenter kan også bli aktuelt.
Ved HCC skjer behandlingen etter individuell vurdering og rådføring med internasjonalt fagmiljø.
Overlevelse (overall survival) for HCC er rundt 50 % etter kirurgisk reseksjon/transplantasjon.
Oppfølging
Som for de fleste andre solide svulster følges også barn etter leversvulstbehandling i minst 10 år etter avsluttet behandling og gjerne gjennom puberteten. Barna følges dels for å avdekke residiv, men like viktig er oppfølging med tanke på senskader etter behandlingen.
De to første årene kommer barna til radiologisk undersøkelse hver tredje-fjerde måned. De fleste greier seg med ultralyd abdomen og røntgen thorax, mens noen må ta MR/CT. Man forsøker å unngå CT i kontrolloppfølging grunnet stråledosen. Etter 2 år blir det gradvis lengre intervall mellom kontrolltimene, og vanligvis avsluttes radiologisk oppfølging etter 5 år.
Barn som har fått store kumulative doser kjemoterapi (Cisplatin, Doxorubicin) som kan skade hjerte, nyre eller hørsel får relevante undersøkelser 1, 5 og 10 år etter behandlingsavslutting (kardiotox, hørselstest, GFR).
Kirurgisk behandling av maligne leversvulster
Behandlingen av leverkreft foregår i tett samarbeid mellom barneonkologer, barnekirurger og transplantasjonskirurger. Komplett fjerning av tumor med frie reseksjonsrender er kritisk for langtidsoverlevelse for både hepatoblastom og hepatocellulært carcinom. Leveren har stor regenerasjonsevne, og reseksjoner opp mot 80 % kan tolereres. Hepatoblastomer vil oftest være kjemosensitive, og preoperativ kjemoterapi kan gjøre kirurgien tryggere. Noen svulster kan ved diagnosetidspunktet gi inntrykk av å involvere aller fire segmenter i henhold til Pretext-inndeling. Etter kjemoterapi vil man oftest se at svulsten kan fjernes. Ved multifokale svulster og Pretext IV-svulster vil levertransplantasjon være aktuelt. For enkelte sentralt beliggende svulster som ikke kan fjernes, kan også levertransplantasjon være et alternativ. Komplikasjoner relatert til leverreseksjoner er blødning, gallegangsskader, karskade, luftemboli og leversvikt.
Germinalcellesvulster utenfor CNS
Germinalcellesvulstene (GCT) er en svært heterogen gruppe svulster som varierer betydelig i klinisk presentasjon, lokalisasjon, histologi og biologi. Hos ungdom og unge voksne er de oftest lokalisert i gonadene. I barnealder derimot oppstår de oftest ekstragonadalt i midtlinjen, f eks sacrococcygealt eller i fremre mediastinum. GCT diagnostiseres i alle aldersgrupper fra fosterlivet og opp gjennom oppveksten og står for 3–4 % av krefttilfellene hos barn < 15 år. Modne teratomer kan være underrapportert i slike statistikker (Gobel et al., 1998; Gobel et al., 2000).
Histologi
Svært varierende mikroskopiske bilde. Det ses benigne tumorer, såkalte modne teratomer som kan bestå av mange ulike vevstyper. Det ses også svulster med større eller mindre malignt potensiale, såkalte maligne germinalcellesvulster (MGCT).
Revised Histological Classification of Germ Cell Tumours
- Germinoma
(1) Intratubular
(2) Invasive - Teratoma
a. Mature
b. Immature
c. Malignant teratoma (Teratoma with non germ cell malignant component) - Embryonal carcinoma
- Yolk sac tumour
- Choriocarcinoma
- Gonadoblastoma
- Mixed malignant germ cell tumour
Genetikk
Tilstander som disponerer: Klinefelter syndrom disponerer for mediastinal MGCT.
Ovariale GCT’er viser en signifikant assosiasjon med konstitusjonelle forandringer i kjønnskromosomer som f.eks ved Turner syndrom.
Diagnose
GCT presenterer seg oftest som en asymptomatisk, stor, plasskrevende oppfylning som kan presse på andre organer og gjennom det gi symptomer, for eksempel obstruksjon i luftveiene, vena cava-syndrom, obstipasjon ved bekkensvulster osv. GCT’er skiller ofte ut tumormarkører (AFP og/eller β‑HCG) avhengig av histologisk differensiering. Mønsteret av disse er med på å gi en klinisk diagnose. Husk at AFP normalt er svært høyt i nyfødtperioden og først er nede i «voksne verdier» ved 1–2 års alder. (Mer om AFP, se kapitlet om leversvulster.)
Utredning
- Grundig anamnese og generell klinisk undersøkelse.
- Blodprøver med tumormarkørpanel (AFP, β-HCG og LD. Evt PLAP (placenta-like alkanin fosfatase) som kan være positiv i serum ved germinomer)
- Evt kromosomanalyse (Klinefelter/Turner o.a.)
- Ultralyd abdomen
- MR (CT) av av aktuelle område hvor tumor er lokaliser og av abdomen
- Rtg thorax og CT thorax
- Urin til katekolaminbestemmelse (differensialdiagnostikk)
Klassifisering og stadieinndeling for MGCT varierer mellom ulike studiegrupper og deres protokoller. Protokollen vi følger, UKCCSG GC III, baserer seg på en vanlig TNM-klassifisering, og behandlingsstrategien deler svulstene i lav-, intermediær- og høyrisiko sykdom avhengig av lokalisasjon, utbredelse og nivåer av tumormarkørsekresjon – for detaljer, se protokoll.
De ulike nasjonale og flernasjonale gruppene har ennå ikke klart å samle seg på samme måte som for en del andre solide svulster. Det er derfor flere grupper med litt ulike studier i Europa samt større studier i regi av COG i USA. Det er imidlertid dannet en International Germ Cell Tumour Group som håper å komme i gang med felles studier hvor også randomiserte spørsmål kan bli besvart.
Vi følger nå den engelske behandlingsprotokollen for GCT – UKCCSG GC III (2005) og Children’s Cancer & Leukaemia Group, Germ Cell Group UK- ‘INTERIM GUIDELINES FOR THE TREATMENT OF EXTRACRANIAL GERM CELL TUMOURS IN CHILDREN AND ADOLESCENTS’ fra 2011. Det gjør også Sverige og Danmark, og vi samarbeider i NOPHOs arbeidsgruppe for GCT. I denne protokollen brukes carboplatin i motsetning til cisplatin som de fleste andre studier benytter. Carboplatin gir mindre skade på hørsel og nyrer enn cisplatin gjør, og carboplatin har vist seg i barnealder å være like effektivt som Cisplatin i de to foregående engelske studiene. Carboplatin gis da i kombinasjon med etoposid og bleomycin (J. R. Mann et al., 2000).
Behandling
Modne teratomer fjernes kirurgisk, men skal følges videre med kontroller (klinisk + ultralyd + markører) da de kan residivere både som modent teratom og som MGCT hvis kirurgien har vært inkomplett. Dette gjelder særlig sacrococcygeale svulster hos spedbarn hvor residivraten er på 10–14 %, og hvor residivene ofte er plommesekksvulster.
Immature teratomer. Kirurgi er hovedbehandlingen og er oftest tilstrekkelig alene. Betydningen av økt AFP ved immature teratomer er kontroversiell, og for tiden anbefales at immature teratomer med AFP> 1000 kU/L ved diagnose behandles som MGCT med kjemoterapi i tillegg til kirurgi. De med AFP < 1000 kU/L skal følges tett med ultralyd hver 3. måned og tumormarkørbestemmelse hver 4. til 6. uke. Ved gjenvekst av tumor anbefales ny kirurgi og deretter kjemoterapi.
Alle GCT’er skal ha målt AFP og HCG og gjennomført full utredning. Hvis metastaser ikke er påvist, skal kirurgi vurderes. Inoperable svulster skal, hvis mulig, biopseres. Mutilerende kirurgi skal unngås. Utsatt kirurgi kan vurderes, men skal fortsatt unngås hvis det medfører skader av betydning. GC II-studien har vist at bare 2 av 45 barn hadde viabelt tumorvev ved utsatt kirurgi etter gjennomført kjemoterapi. De øvrige hadde nekrose/fibrose eller modent/umodent teratom. Slike pasienter bør heller følges tett for å se om utviklingen nødvendiggjør ytterligere behandling.
Lavrisiko MGCT defineres som alle stadium 1 svulster (kfr protokoll) Disse får ingen postoperativ kjemoterapi, men følges tett med ukentlige kontroller til normalisering av tumormarkører og deretter kontroll hver måned.
Intermediær risiko MGCT er alle seminomer/germinomer uansett AFP-verdi, de fleste stadium 2 og 3 svulster med AFP < 10 000 kU/L unntatt thoracale svulster hvor st. 2, 3 og 4 alle er høyrisiko, mens testis st 4 < 5 år er intermediær.
Disse får 4 kurer med kjemoterapi.
Høyrisiko MGCT er alle med AFP > 10 000 kU/L unntatt stadium 1 svulster og untatt testissvulster hos gytter < 5 år. Alle thoracale svulster st 2,3 og 4 inngår i gruppen uavhengig av AFP nivå.
Disse får 6 kurer med kjemoterapi.
Strålebehandling er ikke en integrert del av behandlingsopplegget for ekstrakraniell GCT i barnealder. Men strålebehandling kan være aktuell ved tilbakefall. Dysgerminom i ovariet og seminom i testis er svært strålefølsomme, og strålebehandling kan være aktuelt i utvalgte situasjoner. Men dette vil være etter individuell vurdering hos erfaren stråleonkolog.
Residivbehandling
Protokollen gir også anbefalinger for behandling ved residiv. Kjemoterapien er da vesentlig cisplatinbasert.
Prognose
Prognosen er generelt god. Gjennom de siste 20 år har prognosen for maligne GCT’er bedret seg betydelig. Mye av dette kan tilskrives bedret multimodal tilrettelegging av behandlingen samt ikke minst, introduksjonen av cisplatin i kjemoterapien. Der hvor man kan fjerne tumor radikalt, er overlevelsen nå > 80 %, men enkelte undergrupper som choriocarcinom kan ha dårligere prognose.
Oppfølging
De fleste barn følges i minst 10 år etter avsluttet behandling og gjerne gjennom puberteten. Barna følges dels for å avdekke residiv da disse ofte kan behandles med godt resultat. Like viktig er også oppfølging med tanke på senskader etter behandlingen.
Den første tiden følges de med forhøyede tumormarkører tett for å se at disse faller til normale verdier. Deretter månedlige tumormarkørkontroll i et år, så hver annen måned det andre året og hver 3. måned det tredje året.
De to første årene kommer barna også til radiologisk undersøkelse hver tredje måned. De fleste greier seg med ultralyd abdomen og røntgen thorax, mens noen må ta MR/CT. Man forsøker å unngå CT i kontrolloppfølging grunnet stråledosen. Etter 2 år blir det gradvis lengre intervall mellom kontrolltimene, og vanligvis avsluttes radiologisk oppfølging etter 5 år.
Barn som har fått store kumulative doser kjemoterapi som kan skade hjerte, lunger, nyre eller hørsel får relevante undersøkelser 1, 5 og 10 år etter behandlingsavslutting (kardiotox, lungefunksjonstest, hørselstest, GFR). Man er også oppmerksom på stråleinduserte skader og samarbeider med ortoped, pedagog og andre dersom tegn til stråleskader oppdages. Man er også oppmerksom på endokrinologiske bivirkninger og samarbeider tett med barneendokrinolog, eventuelt gynekolog.
Retinoblastom (RB)
Epidemiologi
Retinoblastom er en sjelden svulst som oppstår i ett eller begge øyne fra umodne retinaceller hos barn. Insidens av RB i Norge er omtrent 1/15 000 levende fødte barn (Gregersen et al., 2016). Det oppdages ca. 4 nye tilfeller per år og det er like mange gutter som jenter som får RB. Selv om RB kan inntreffe i alle aldre, oppstår det oftest før 2 års alderen, og ca. 95 % av tilfellene er diagnostisert før 5 års alder. Opp mot 98 % av alle barn med RB blir helbredet. Dette er et resultat av tidlig diagnostikk og gode behandlingsmuligheter.
Klassifikasjon
Nå klassiffiseres RB hovedsakelig i henhold til den Internasjonale klassifiseringen; ABCDE (Murphree, 2005). Tidligere benyttet man også Reese-Ellsworth (Yousef et al., 2015), og TNM (Rosengren, Monge, & Flage, 1989). Det kan være en fordel å benytte alle tre klassifikasjoner slik at det kan gjøres sammenlikninger med tidligere publiserte materialer.
Veksttyper
Endofytisk: Vokser fra netthinneoverflaten og inn mot eller inn i glasslegemet. Tumorens overflate er oppblåst og dårlig avgrenset med atypiske kar (figur 1).
Eksofytisk: Vokser utover og løfter opp netthinnen til evt. amotio (figur 2).
Diffust infiltrerbar: Dette er en uvanlig form som ofte diagnostiseres sent på grunn av differensialdiagnostiske vanskeligheter. De aller fleste pasientene er eldre enn ved de to andre formene. Tumor er vanligvis ensidig og oppstår spontant (Kim, Abramson, & Dunkel, 2007).
Patogenese, genetikk og etiologi
Retinoblastom skylles mutasjon av begge alleler av RB1 genet, som er lokalisert på kromosom 13 bånd q14 og er et stort gen som omfatter 200 000 basepar (Friend et al., 1986; Kohno, Kitajima, Sasaki, & Takahashi, 2016). RB1 er et tumor-suppressorgen. Pasienter med den arvelige formen har en medfødt germline mutasjon på et av allelene (first hit), som enten kan være nyoppstått eller nedarvet fra foreldrene (10 %). Penetransen ved RB1 mutasjoner er høy, men det finnes tilfeller med nedsatt penetrans (samlet penetrans er ca. 95 %). Tumor oppstår som en somatisk mutasjon i det andre allelet (second hit) (Cook et al., 2017). De fleste av de arvelige tilfellene er multifokale og bilaterale, og gjennomsnittsalderen ved diagnose er 15 mnd. Ved de ikke arvelige, sporadiske tilfellene er tumor oppstått som følge av to somatiske mutasjoner. Disse er oftest unilaterale og unifokale, og gjennomsnittsalderen ved diagnose er ca. 24 mnd.
Ved den arvelige varianten kan det også forekomme tumor i corpus pineale. Dette kalles trilateralt retinoblastom, og MR cerebrum us. er viktig for å utelukke dette.
Symptomer og tegn
Ved undersøkelse av barn, både ved nyfødtscreeningen og på helsestasjonene, bør den minste mistanke om leukokori eller skjeling medføre umiddelbar henvisning til øyelege (Dimaras et al., 2012; Sandven-Thrane, 2012). De vanligste debutsymptomene er:
- Leukokori (hvit refleks): 56–69 %
- Strabisme (skjeling): 20–35 %
- Nedsatt syn
- Glaukom
- Rødt øye
- Uveitter
- Unilateral mydriasis
Utredning
Øyeundersøkelse i våken tilstand: Sjekk av visus, synsfelt, skjeling, pupillerefleks og øyebevegelse.
Øyeundersøkelse i full narkose: Undersøkelse av øyebunnen i maksimal mydriasis og med oraimpresjon. Bruk av ultralyd for måling av tumorstørrelse, vurdere innhold av kalk og ev. gjennomvekst av øyeveggen. Klinisk foto av øyebunnen. Biopsi er ikke anbefalt p.g.a. risiko for sekundær spredning.
Blodprøver: Hb, leukocytter med differesialtelling, trombocytter, natrium, kalium, kreatinin, ASAT, ALAT, LD, CRP, leverfunksjonsprøver, serologiske undersøkelser for CVM, EBV og Varicella og prøver til genetikk.
Full somatisk undersøkelse.
MR orbita og cerebrum: Framstiller godt tumor fra vevet omkring. Ser etter mulig ekstraokulær vekst og corpus pinealesvulster. Tumorvevet fremstilles som moderat hyperintensivt på T1 og hypointensivt på T2 bilder. MR kan også skille mellom tumorvev, blødning og eksudat.
Barnet og familien henvises til genetisk samtale/veiledning.
Ev. CT orbita: Fremstiller kalk i tumor, god på påvisning av vekst utenfor øyet.
Ved ev. enukleasjon: Ta ferskt tumorvev til mutasjonsanalyse før formalinfiksering.
Behandling
Innledning
Både diagnostikk og behandling av retinoblastom er multimodal og omfattende og skal gis ved sykehus med spesialkompetanse i slik behandling.
Øyeavdelingen og Barneavdeling for kreft og blodsykdommer ved Oslo Universitetssykehus har landsfunksjon for retinoblastom. Alle retinoblastomene utredes og behandles primært her. Alle oftalmologiske kontroller under behandlingen gjøres her.
Små svulster kan behandles med lokalbehandling, som kryobehandling, laser behandling eller stråleplate, utført ved øyelege. Ved større svulster kan det være aktuelt med kjemoterapi, og i noen tilfeller må man fjerne øyet (enukleasjon). Ekstern stråling brukes sjelden i dag.
Om det skal gis konvenskonell, intravenøs kjemoterapi, kan dette gis på lokalsykehus med kompetanse på kjemoterapi. Mellom kurene kan barnet gjerne være hjemme, men må da ha tett kontakt med lokalsykehuset for blodprøvekontroller. Støttebehandling i form av transfusjoner og eventuell antibiotikabehandling, gis på lokalsykehuset etter vanlige retningslinjer.
Kjemoterapi
Ved behov for kjemoterapi, er primærbehandlingen nå ofte cytostatika (Melfalan) direkte inn i a. ophthalmica (IAM). Dette har vist goderesultater hos de fleste (Radros et al., 2018). I tillegg unngår man bivirkningene som ved systemisk behandling. Denne behandlingen brukes som volumreduserende tiltak før annen behandling (Shields et al., 2004; Shields, Meadows, Shields, Carvalho, & Smith, 2001). Nødvendigheten av tilleggsbehandling vurderes ved øyeundersøkelse i narkose i tilslutning til hver 2. kur. Om ikke IAM behandling er aktuelt eller er kontraindisert (for eksempel hos de aller minste barna), kan det gis systemisk kjemoterpi med Vincristin, Carboplatin og Etoposid med 3 ukers mellomrom. Det gis vanligvis 3–6 kurer.
Kryobehandling
Vevet fryses til –70 °C. Benyttes ved mindre perifere svulster (mindre enn 6 mm i diameter og 3 mm høyde).
Laserbehandling
Det finnes ulike typer laser som benyttes for å ødelegge blodforsyningen til tumor. Benyttes ved små svulster (mindre enn 4–7 mm i diameter og 3 mm høyde) særlig de som ligger langt bak i øyet. Kan også gis i tilslutning til kjemoterapi primært, eller ved små residiv.
Strålebehandling
Retinoblastom er en strålefølsom tumor (Kim et al., 2007; Swanson et al., 2011)
Lokal strålebehandling med plate (Ru-106 (ruthenium) eller I-125 (jod)). Disse avgir betastråler respektive lavenergetiske gammastråler.
Indikasjoner:
Tumor < 15 mm i diameter og 3–7 mm tykk.
Tumores som er minst 2 mm fra n. opticus eller macula.
Tumores med lokal infiltratrasjon i glassvæsken.
Turmores som ikke har svart på annen behandling eller ved residiv.
Dosen er 35–40 Gy mot tumors apeks.
Ekstern strålebehandling
Indikasjon
Ved residiv hvor man vil bevare et seende siste øye, hvor annen behandling ikke har virket, eller ved spredning ut i glasslegemet.
Dosen er 35–40 Gy, 1,5–2 Gy 5 dager per uke i 5 uker.
Om det er aktuelt med strålebehandling, gis dette i form av protonbestråling som har kortere strålerekkevidde og derfor ikke skader tilstøtende vev like mye som fotoner.
Enukleasjon
Benyttes enten når svulsten ved diagnosen er så stor at det ikke er mulig å bevare visus, ved residiv man ikke får under kontroll, eller ved tegn til metastaser. Ved enukleasjon er det viktig at man får med seg så mye av n.opticus som mulig. Dersom det ved histologisk undersøkelse av det fjernede øye finnes tegn til gjennomvekst i n. opticus, eller ved tegn til infiltrasjon gjennom sklera, er det nødvendig med kjemoterapi eller strålebehandling (Nordiske retningslinjer for retinoblastom, 2016).
Prognose
Overlevelsen av RB er 98 %. Lokalt residiv er vanlig, men kommer svært sjelden etter at barnet har fylt 3 år.
Prognostiske faktorer mhp residiv:
Tumors størrelse og utbredelse.
Eventuelt forhøyet intraokulært trykk ved diagnosen.
Alder ved diagnose. Prognosen med henblikk på residiv er bedre ved diagnose mellom 2 og 7 år enn for pasienter som diagnostiseres tidligere eller senere.
Risiko for nye retinale tumores er (Abramson, Greenfield, & Ellsworth, 1992)
58 % hvis diagnosen stilles < 3 mnd alder
45 % hvis diagnosen stilles mellom 3 og 6 mnd alder
14 % hvis diagnosen stilles > 6 mnd alder
De som har arvelig retinoblastom, har større risiko for å utvikle sekundære maligniteter, dvs. sarkom i ben og bløtvev og kutane melanomer (Kleinerman et al., 2005; Kleinerman et al., 2012; Turaka, Shields, Meadows, & Leahey, 2012; Wong et al., 2014). 70 % av tilfellene oppstår i hodet (innen strålefeltet) ved ekstern strålebehandling Dette er avhengig av dose/respons og alder. Disse tumortypene andre steder i kroppen er ikke relatert til strålebehandling, men til mutasjonene i RB1-genet.
Utvikling av ev. komplikasjoner i øyet
Fra gjenværende tumor og arrvev
Vaskulære anomalier
Blødninger
Forandringer på retina
Glaukomutvikling
Ofte utvikling av pussdannelse ved enukleasjon.
Forandringer etter strålebehandling
Kosmetiske
Sekundær atrofi av orbita
Atrofi av fett og bindevev – øyet blir innsunket
Stråleretinopati og opticusnevropati
Utvikling av katarakt
Ofte store plager med tørre øyne, trenger kunstig tårevæske
Videre kontroller
Barn som er behandlet for RB bør gå til hyppige kontroller både hos pediater og oftalmolog med henblikk på:
Residiv
Ny cancer, primær eller sekundær (i strålefeltet)
De bør også følges opp på lokal BUP ved behov.
Øyelegekontroll:
Barn som har gjennomført behandling for retinoblastom bør kontrolleres:
Hver 2. mnd et år etter siste behandling. Deretter individuell nedtrapping. Avhengig av antatt arvelighet og gjenværende tumorforandringer i øyet/øynene skal noen ha kontroll hos øyelege hele livet.
Barn til foreldre med bilateralt retinoblastom bør kontrolleres:
Så raskt som mulig etter fødsel (innen maks 2–3uker)
Hver 2.–3. mnd til 2 års alder.
Hver 4. mnd til 4 års alder
Hver 6. mnd til 7–8 års alder
Søsken til barn med retinoblastom og barn til foreldre med unilateralt retinoblastom bør kontrolleres:
Hver 2.–3. mnd første året.
Hver 4. mnd til 4 års alder.
Hver 6. mnd til 7–8 års alder
Sjeldne svulster
Sist faglig oppdatert: 26.05.2020
I tillegg til de svulstene som her er omtalt, finnes det en rekke andre svulsttyper som forekommer svært sjelden. Dette gjelder dels svulsttyper som er mer eller mindre spesifikke for barnealder, dels mer «voksne» svulster. Felles for disse ekstremt sjeldne svulsttypene er at det ikke finnes spesifikke behandlingsprotokoller for barn, og man må som regel i hvert enkelt tilfelle skreddersy en behandling ut fra histologisk type, barnets alder og egne eller andres erfaringer med lignende svulsttyper.
Støttebehandling og rettigheter
Medisinsk støttebehandling
Sist faglig oppdatert: 26.05.2020
Når barn får kreft, får de plager på grunn av både sykdommen og behandlingen. Kanskje har de andre sykdommer i tillegg. Derfor er det er viktig å ivareta «hele» barnet for å bevare en god livskvalitet. Foreldre og barn trenger tilpasset og tilrettelagt informasjon om sykdom og bivirkninger. Informasjonen må tilpasses barnets alder og foreldre og barns kulturelle og språklige tilhørighet. Det er viktig å involvere støttepersonell som fastlege, sykepleier, fysioterapeut, dietetiker, tannpleier, sosionom, prest, skole og barnehage etter behov. De eldste barna vil ofte ha andre informasjonsbehov underveis, som for eksempel informasjon om seksualitet og kreft, og de kan ha behov for diskusjon rundt eksistensielle problemer.
Helsesøster bør involveres om familien ønsker det. Det er viktig med samtykke før eksterne involveres.
Symptomer som oppstår underveis, kan være uttrykk for mer eller mindre alvorlige sykdoms – eller behandlingsrelaterte komplikasjoner. Det dreier seg om anemi, infeksjoner og tumorlysesyndrom. I hvilken grad slike komplikasjoner oppstår, avspeiler intensiteten av cellegiftbehandlingen, kreftsykdommen, barnets tilstand på diagnosetidspunkt og genetiske forhold.
Støttebehandling omfatter symptomlindring med antiemetika, analgetika, steroider, H2‑reseptorantagonister, protonpumpehemmere, benzodiazepiner og laksantia. Det gis ofte antibiotika, blodprodukter, væske og elektrolytter, hematopoietiske vekstfaktorer og noen ganger antiepileptika og antikoagulantia. Barnets ernæringstilstand bør tidlig ivaretas, og bruk av nasogastrisk sonde, perkutan gastrostomi eller parenteral ernæring er vanlig.
Profylaktisk antimikrobiellbehandling
Behandling av barn med cellegift, stråleterapi og eventuell organtransplantasjon øker risikoen for infeksjoner. Spesielt behandling med langvarig myelosuppresjon med slimhinneaffeksjon, sentrale katetre og langvarig sykehusopphold, hvor barnet er utsatt for nosokomielle organismer, bidrar til å øke infeksjonsrisikoen. Det stilles derfor store krav til behandlende avdeling, som må ha rutiner for å redusere smittespredning og for opplæring av barn og familie i profylaktisk munnhygiene og alminnelige hygieniske prinsipper. Retningslinjer for infeksjonsprofylakse i forbindelse med kreftbehandling finnes ofte beskrevet i dagens protokoller som det henvises til på https://www.nopho.net/ og https://www.barnekreftportalen.no/. De hyppigste agens som krever profylaktisk behandling er Pneumocystis jirovecii, sopp og Varizella zoster virus (VZV). Stamcelletransplantasjon er en spesiell situasjon med langvarig utslått immunforsvar. Barn i denne situasjonen kan utvikle kronisk GHVD og kan ha forlenget behov for profylaktisk behandling mot Pneumocystis jirovecii, sopp og virus (CMV, HSV og VZV). Indikasjon og dosering av antibiotika er beskrevet i Barnelegeforeningens generelle veileder og akuttveileder for pediatri (Akuttveileder i pediatri [e-bok], 2013; Generell veileder i pediatri [e-bok], 2012).
Retningslinjer for bruk av G-CSF
G-CSF brukes som primærprofylakse for å kunne øke doseintensiten av cellegiftbehandlingen. Dette er aktuelt ved for eksempel HR-nevroblastom, som sekundærprofylakse hos pasienter som tidligere har utviklet nøytropen feber, og hos kritisk syke barn med langvarig nøytropen feber etter cellegiftbehandling. G-CSF (filgrastim, Neupogen®) doseres for barn med 5 µg/kg/dag s.c (NB høyere dose i forkant av stamcellehøsting, vanligvis 10 µg/kg/dag). Varighet av behandlingen er inntil nøytrofile > 5 x 109/L en dag eller > 1 x 109/L i tre dager eller i henhold til protokoll.
Transfusjon
Anemi ved malign sykdom er ofte multifaktoriell. I de fleste tilfeller er kjemoterapien årsak, men også hemning av erytropoiesen, benmargsmetastasering, blødning og mangelfull ernæring spiller en rolle. Det er per i dag ingen konsensus om indikasjon for transfusjon av SAGMAN-erytrocyttkonsentrat i kreftbehandling, og beslutningen tas etter vurdering av barnets diagnose, allmenntilstand og forventede forløp. Den kliniske beslutningsterskel for transfusjon vil endres under behandlingsforløpet, hvor barnet tilvenner seg en lavere hemoglobinkonsentrasjon. På diagnosetidspunkt kan barnet ha svært lav hemoglobinkonsentrasjon (< 5 g/dL), og hyppige, mindre transfusjoner er aktuelt. I noen tilfeller, som ved nyoppdaget leukemi, kan det være en fordel med lav hemoglobinkonsentrasjon for å begrense viskositeten og risikoen for leukostase (se også avsnitt "Hyperleukocytose og leukostase" i kapittel Akutte onkologiske tilstander). En ofte brukt grense for blodtransfusjon er 8 g/dL, bortsett fra perioder med stråleterapi hvor en grense på 10 g/dL anbefales. Indikasjonsstillingen må imidlertid vurderes individuelt ut fra barnets kliniske tilstand og om man kan forvente snarlig spontan økning (retikulocytter). Dosering av SAGMAN-erytrocyttkonsentrat: (Ønsket hemoglobinkonsentrasjon (g/dL)– aktuell hemoglobinkonsentrasjon) x vekt x 3. Vanligvis blir dette 10–20 mL/kg kroppsvekt.
Før planlagt stamcelletransplantasjon skal alle blodprodukter bestråles fra ca. 1 måned før til ca. 12 måneder etter. Se Barnelegeforeningens generelle veileder og akuttveileder for pediatri (Akuttveileder i pediatri [e-bok], 2013; Generell veileder i pediatri [e-bok], 2012).
Indikasjon for trombocytt-transfusjon ved trombocytopeni og redusert produksjon er oppsummert i tabellen nedenfor.
Dosering er: Barn < 25 kg: 15 ml/kg kroppsvekt. Barn > 25 kg: 1–2 enheter. Trombocyttene gis over 20–40 minutter.
| < 10 x 109/L | < 20 x 109/L | 20–40 x 109/L | < 50 x 109/L |
| Som hovedregel gis alltid trc |
| DIC i induksjonsfasen med uttalt hyperleukocytose | Ved diagnostisk lumbalpunksjon for ALL anbefales > 50 |
Ved blødning og trombocytopeni skal det alltid gis trombocytter.
Det henvises for øvrig til Generell veileder for pediatri (Generell veileder i pediatri [e-bok], 2012) om indikasjon for leukocyttfiltrering og bestråling av blodprodukter generelt samt referanse (Gibson et al., 2004).
Smertebehandling
Barn med kreft har ofte smerter på diagnosetidspunkt, og det er viktig å gi optimal smertelindring, både for at barnet skal ha det best mulig og for at barnet skal bevare tilliten til behandler. I de fleste tilfeller avtar behovet raskt etter start av cellegiftbehandling. For både leukemi, solide svulster og CNS-svulster vil det være perioder hvor barnet trenger intensivering av smertebehandlingen. Det er alltid viktig å avklare smerteårsak, smerte utbredelse, type smerter (somatisk og/eller visceral nociseptiv- eller nevrogen smerte), gradere smerten og avklare grad av engstelse som vil kunne forsterke smerten. Tidlig introduksjon av smerteskala tilpasset barnets alder kan hjelpe barnet å gradere smertene og behandler å optimalisere behandlingen. Smertelindring i terminalfasen er en spesialistoppgave og igangsettes av behandlende onkologisk avdeling (se eget kapitel om palliativ behandling).

Vanlige prinsipper for smertebehandling kan brukes. Man starter med paracetamol som kan kombineres med NSAIDs (ibuprofen), og rask opptrapping med opiater. Forsiktighet med NSAIDs dersom barnet har redusert eller truende nyrefunksjon, får nefrotoksiske medikamenter/cellegift, ved moderat til alvorlig trombocytopeni og ved kjent allergi. For å finne barnets døgnbehov startes opiatbehandling med hurtigvirkende preparater, eventuelt med intravenøs tilførsel ved alvorlige smerter eller ved problemer med tablettinntak. Døgndosen kan senere gis som langtidsvirkende preparat, men det må alltid være mulighet for hurtigvirkende opiater ved gjennombruddssmerter og rask oppjustering av doser ved manglende effekt. Ved intensiv smertebehandling økes risikoen for obstipasjon, og bruk av laxantia anbefales. De fleste barn vil være innlagt i slike situasjoner, og behandlingen styres av smerteteam. For supplerende informasjon henvises det til: www.childcancerpain.org, www.legemiddelhandboka.no og generell veileder i Pediatri
Ikke-medikamentell smertelindring bør vektlegges: En barnepsykiater og psykolog skal i dag ha kompetanse på en rekke ikke-medikamentelle symptomlindrende teknikker; bl.a. ift smerte og kvalme. Spesielt gjelder dette ved tillegg av angst og uro (som nesten alltid vil være til stede). Eksempler på psykologiske intervensjoner som ledd i smertelindring er; avledning, narrativer, avspenning og pusteteknikker, eksternaliseringsteknikker, hypnoterapi, korttidsterapi (CBT) m.m. Et viktig ledd i smertelindring av barn, er god ivaretagelse/trygging av foreldre/pårørende.
Psykososiale tiltak
Sist faglig oppdatert: 26.05.2020
Innledning
Målet for psykososiale tiltak når barn får en kreftsykdom, er å fremme mestring for barnet og dets betydningsfulle nære (familien), samt å redusere risiko for utvikling av psykisk lidelse. Iblant også å behandle psykisk lidelse hos barnet. Det er mange forskningsrapporter som omtaler incidens og prevalens av ulike psykologiske belastninger/uttrykk i sammenheng med somatisk sykdom (for en oversikt, se ref (Rutter's child and adolescent psychiatry, 2008)). Sammenfatningsvis kan man si at mange barn og unge sliter med angst, kognitive senskader (bla svekket hukommelse og nedsatt psykomotorisk tempo, lærevansker), depresjon og posttraumatiske symptomer i etterkant av alvorlig somatisk sykdom (der kreftsykdommer utgjør en av de største sykdomsgruppene).
Barns tilpasning til kreftsykdom
Hvilke psykososiale tiltak som er aktuelle å iverksette, avhenger av en mengde faktorer, der de faktorene som påvirker barns tilpasning til sykdom og behandling, trolig er de viktigste (for utdypende informasjon, se ref (Reinfjell, Diseth, & Vikan, 2007)). De viktigste faktorene for barns tilpasning til kronisk og/eller alvorlig sykdom er:
- Karakteristika ved sykdommen (i hvilken grad den er livstruende og begrenser bevegelse og sosiale aktiviteter eller opplevelser)
- Situasjonelle faktorer som stressorer i barnets behandlingshverdag (behandlingsprosedyrer, bivirkninger og komplikasjoner).
- Karakteristika ved barnet (alder, kjønn, personlighet eller mestringsstil og tidligere erfaringer).
- Karakteristika ved familien (problemløsningsevne eller kommunikasjonsferdigheter, samt grad av åpenhet i familien), og sosial støtte fra venner og familie.
Negative følelser som sinne, skyld, skam, ensomhet, apati, bitterhet og forvirring, observeres ofte hos barn med kreft. Dette kan bidra til isolasjon. En finner ofte symptomer på depresjon hos barn med kreft. Mange er redde for døden og for at sykdommen kan slå til igjen. Mestringsstrategier benyttes for å minske eller eliminere problemene som er nevnt ovenfor: Å søke informasjon, støtte og trøst, lete etter årsaker til hendelser, forandre situasjonen, benekte og unngå, impulsiv handling, kognitiv restrukturering og akseptere situasjonen.
Strategiene benyttes for å redusere følelsen av usikkerhet, for å oppnå og beholde en form for kontroll over situasjonen, for å opprettholde og beskytte selvaktelsen og for å redusere negative følelser (Van Dongen-Melman et al., 1995). Hvordan kan disse utfordringene imøtekommes? I det følgende blir aktuelle psykososiale tiltak presentert:
Ulike psykososiale tiltak
- Informasjonssamtaler og betydningen av åpenhet
- Støttesamtaler (krisepsykologiske- og mestringsfremmende intervensjoner)
- Hjelp til å mestre somatisk behandling (smertelindring, sprøyteskrekk m.m.)
- Likemannsarbeid/Foreldrekontakttjeneste
- Sorgarbeid
- Samtalegruppe (pasient, søsken, foreldre)
- Indirekte arbeid (veiledning, tverrfaglige møter m.m.)
- Nevropsykologisk utredning
- Videre oppfølging
Informasjonssamtaler og betydningen av åpenhet
For barn der det er mistanke om, eller konstatert, kreftdiagnose, bør personer med psykisk helsefaglig kompetanse trekkes inn allerede under diagnostiseringsprosessen. Dette for å bistå med krisepsykologiske intervensjoner overfor familien som helhet, samt for å gjøre den medisinske informasjonen mest mulig forståelig for barnet/barna i en familiesetting. I denne prosessen vil det være sentralt å få tak i barnets (og familiens) forståelse av situasjonen, aktuelle bekymringer og mestringsstrategier. Foreldreveiledning kan være et viktig innslag gjennom hele behandlingsprosessen (med fokus på hvordan foreldre kan snakke med sine barn om sykdommen og behandlingen, hvordan de kan skjerme og beskytte barnet (f.eks ved å si stopp når barnet ikke er klar for en gitt prosedyre), og hvordan bearbeide hendelser i etterkant (eks ved hjelp av Kiwanis dukker). Det er viktig å tilby hyppige oppfølgingssamtaler de første ukene, for så å kartlegge behov for videre psykososial oppfølging. Der barnet er innlagt ved et sykehus er det naturlig at denne oppfølgingen skjer på sykehuset i nært samarbeid med somatiker. I senere faser, samt ved kortere sykehusopphold, vil det være å foretrekke at denne oppfølgingen skjer lokalt (helsestasjon, familiekontor, andre lokale helse og omsorgstjenester). Støttepersoner i Barnekreftforeningen vil også kunne være en stor ressurs i denne fasen.
Det er nå en rådende oppfattning at ærlig og åpen kommunikasjon er avgjørende for å fremme psykisk helse hos barn, og for å bringe fortrolighet med behandlingen. En rekke studier har dokumentert at innhenting av nøyaktig informasjon er av stor betydning for å redusere usikkerhetsfølelse og tendenser til depresjon (Van Dongen-Melman et al., 1995). Dette vil noen ganger medføre behov for tilrettelagt kommunikasjon via tolk. Det er viktig at det holdes nye informasjonssamtaler med lege og psykisk helsepersonale når behandlingen går over i en ny fase, når det skjer store forandringer i sykdomsutviklingen, samt med jevne mellomrom (ettersom barnet får nye erfaringer, og modnes).
Informasjonssamtaler må tilrettelegges og det må vurderes å bestille tolk når barnet eller dets omsorgsgivere/foreldrene har begrensede norskkunnskaper. Eventuell skriftlig informasjon må vurderes oversatt til aktuelle språk for å sikre at viktig informasjon blir forstått.
Personer utenfor familien behøver også informasjon. Eksempelvis barnets (og evt søskens) skole. Det er viktig at dette temaet tas opp med familien allerede den første tiden, slik at familien kan være med på å bestemme hvem som skal få hvilken informasjon av hvem, og på hvilken måte. Kreftsykepleier kan være en viktig ressurs i dette arbeidet.
Å snakke åpent om døden er for mange vanskelig og tabubelagt; det ligger ofte en stor gjensidig beskyttelsestrang mellom barn og voksne, spesielt i den nærmeste familie. Studier indikerer at det å samtale om døden med sitt døende barn kan hjelpe barnet i den vanskelige sluttfasen av livet og foreldrene i sin sorgbearbeidelse etterpå. Fagpersoner må tørre å være til stede i siste fase når et barn skal dø. Det innebærer å være emosjonelt til stede for barnet og familien. En viktig forutsetning er at helsepersonell våger å gå inn i seg selv, kjenne på og reflektere over egne holdninger til døden. Alle som arbeider med alvorlig syke barn bør få faglig veiledning.
Støttesamtaler (krisepsykologiske- og mestringsfremmende intervensjoner)
Alle familier som har barn i behandling for kreft, skal tilbys samtale med psykisk helsepersonale. Når noen i familien har begrensede norskkunnskaper må samtalen tilrettelegges via tolk. Slike samtaler bedrer barnets og familiens mestring av situasjonen, samt virker preventivt for etterreaksjoner (utvikling av psykisk lidelse og negative psykologiske reaksjoner). Hvor hyppig, med hvilke familiemedlemmer, og med hvilket fokus avgjøres ifht de fire faktorene for barns tilpasning til kronisk og/eller alvorlig sykdom, slik nevnt ovenfor, familiens ønske, samt tilgjengelige ressurser innen psykisk helsevern (sykehusets CL-psykiatri- team2, lokal BUP3, ulike lokale/1. linje tilbud).
Alvorlig sykdom hos barn kan forstyrre samspillet og den affektive dialogen mellom barnet og den voksne, og dermed være en risikofaktor for barnets utvikling. Et viktig spørsmål er hva familien trenger av hjelpetiltak for å unngå/begrense dette. Individuelle samtaletilbud til foreldre, så vel som familieterapi, kan være en viktig støtte. Foreldre kan ha opplevd tidligere kriser som reaktiveres når barnet får kreft. Det kan være tilleggsvansker i familien utover barnets kreftsykdom, for eksempel psykiske lidelser, og enkelte vil i større grad trenge oppfølging (ved barneavdelingen, henvisning til lokal DPS4, eller familievern5. Men kreftsykdom hos et barn er i seg selv en risikofaktor og kan true samspillet. Enkelte studier indikerer også posttraumatisk stresslidelse (PTSD) hos foreldre til barn med kreft etter at barnet er ferdigbehandlet (Van Dongen-Melman et al., 1995).
Det vil være nyttig å innhente en del anamnestiske data fra familien. Noen viktige punkter vil være tidligere erfaring med sykdom, skade og andre alvorlige livshendelser, og mestring av disse situasjonene. I Rutter et al. (Rutter's child and adolescent psychiatry, 2008) blir poenget med å høre foreldrenes beskrivelse/opplevelse av hendelsesforløpet så langt, understreket. En slik gjennomgang vil kunne skape bedre oversikt/kontinuitet, den vil kunne gi rom for nye betraktinger/forståelse av situasjonen og dermed nye mestringsmuligheter. Ulike behov/mestringsstiler kan også komme frem, noe som kan anerkjennes og medføre større toleranse overfor andre familiemedlemmers håndtering. Den vil også kunne si noe om hva familien opplever som god hjelp.
Et annet fokus som bør ivaretas i samtaler med psykisk helsepersonell, er fokus på døgnvaner, det vil si naturlige, helsefremmende gjøremål som søvn, spising, fysisk aktivitet, frisk luft, impulser utenfra sykehuset, med mer. Dette er selvsagt spesielt viktig ettersom behandling av kreftsykdom oftest strekker seg over lang tid.
Hjelp til å mestre somatisk behandling (smertelindring, sprøyteskrekk m.m.)
Behandling for kreftsykdom innebærer mange angstutløsende og smertefulle prosedyrer, i tillegg til smertefulle bivirkninger og sykdomsopplevelse. For mange barn (alene eller i en familiesetting) vil det være nyttig å lære nye mestringsstrategier for dette. Psykisk helsepersonell kan være nyttig samarbeidspartner med tanke på å lære avledningsteknikker og avslapning. Psykolog og lege (med relevant kursing/tilleggsutdannelse) kan også utøve hypnose og hypnoterapi. I denne sammenheng har hypnose/hypnoterapi vist seg å være spesielt virkningsfullt som smertelindring og for å mestre angstutløsende/smertefulle prosedyrer.
Likemannsarbeid/Foreldrekontakttjeneste
I Barnekreftforeningen er det en landsdekkende familiekontakttjeneste som består av foreldre som selv har, eller har hatt, barn med kreft.
Foreldrekontakttjenesten består av en koordinator og flere foreldrekontakter. Dette kalles likemannsarbeid, og er en av Barnekreftforeningens grunnpillarer. Når et nytt medlem melder seg inn, vil de i løpet av de nærmeste dagene bli kontaktet av foreldrekontakttjenesten. Dette er ikke medisinske eksperter, men foreldre som selv har vært gjennom det de nye familiene nå står overfor.
Foreldrekontaktene sitter inne med følelsesmessige erfaringer og, ikke minst, svar på praktiske spørsmål som ofte dukker opp i denne første fasen. De aller fleste opplever familiekontakten som en særdeles god støtte i en vanskelig tid (Foreldrekontakttjenesten, 2013).
Sorgarbeid
Det vil være av stor betydning å følge opp familier som har mistet barn etter kreftsykdom. Dette kan gjøres av sykehusets CL-team, i samtalegrupper (søskengrupper, foreldergrupper – et tilbud som finnes på flere større sykehus, samt i 1. linjetjenesten på flere steder) eller gjennom å ta kontakt med lokalt familievern/BUP. En generell anbefaling er at det finnes et sorgtilbud tilgjengelig ved hvert sykehus.
Samtalegruppe (pasient, søsken, foreldre)
For de fleste familier har det stor nytteverdi å møte andre familier som er i tilsvarende situasjon. Det å få utveksle erfaringer, og ikke minst å få fortelle sin historie til noen som kan sette seg inn i deres opplevelser, minsker opplevelsen av alenehet, samt gjør egne tanker og følelser mer aksepterbare og forståelige. Dette kan selvsagt skje av seg selv, men oftest vil det å organisere et slikt tilbud, hvor psykisk helsepersonell har en fasilitatorrolle, ha større effekt. Det vil som oftest være hensiktsmessig å ha egne foreldregrupper, søskengrupper (inndelt etter alder) og eventuelt pasientgrupper. Behov for tilrettelegging via tolk må vurderes.
Indirekte arbeid
En viktig del for å imøtekomme behov innen psykisk helse for denne pasientgruppen er at psykisk helsepersonell deltar i ulike behandlingsmøter. Dette inkluderer previsitt og den aktuelle avdelingens ulike tverrfaglige møtepunkt. I tillegg kan det være aktuelt med gruppeveiledning (av sykehuspersonalet) og undervisning. Alle disse punktene er, foruten å ivareta psykisk helse hos den enkelte pasient og dennes familie, viktig for å øke behandlingspersonalets kompetanse innen psykisk helse – barn og unge.
Nevropsykologisk utredning og oppfølging
For barn med hjernesvulst finnes det et nasjonalt oppfølgingsprogram som også inkluderer jevnlig nevropsykologisk testing. Det er utarbeidet prosedyrer for hvor hyppig barna skal undersøkes og hvilke instrumenter som skal benyttes. Alle barn i landet følges opp ved ett av fire sykehus når det gjelder nevropsykologi: Tromsø, St. Olav, Haukeland og OUS (Ullevål og Rikshospitalet). Ved alvorlige kognitive sekveler viderehenviser barnehabiliteringen til spesialpedagogisk oppfølging fra et av Statpeds kompetansesentra6.
Når det gjelder barn med andre typer kreft, er det ikke noe automatikk i at disse barnegruppene henvises til kognitiv vurdering, verken i regi av barnehabiliteringstjenestene eller PPT7. Selv om ikke kreftsykdommen eller behandlingen får noen direkte konsekvens for kognitiv fungering, er det slik at en del barn mister skolegang på grunn av mange sykehusinnleggelser.
Sykehusskolen kan ikke kompensere for dette fullt ut, da barn ofte har nedsatt allmenntilstand når de er innlagt, og det kan således være behov for særlige tiltak. Barn som får CNS-bestråling som ledd i behandlingen for leukemi, kan få kognitive sekveler på linje med barn med hjernesvulst. Selv om det altså ikke finnes noen oppfølgingsprosedyre for denne barnegruppen, bør man være oppmerksom på om barnet bør henvises til den lokale barnehabiliteringen.
Videre oppfølging
Å legge en god plan for videre oppfølging vil være av største viktighet. Her er det spesielt viktig å trekke inn lokale tverrfaglige ressurser (skole, ppt, bup, fysioterapi, fastlege og andre instanser etter behov). Langtidskonsekvenser av å ha vært igjennom kreftbehandling kan være mange, blant annet fatigue, smerter og konsentrasjonsvansker. Tett og god oppfølging av disse og andre symptomer vil derfor kunne være sentralt i lang tid etter avsluttet kreftbehandling.
Relevante instanser og samarbeidspartnere på sykehus
- Lekerom/aktivitetsrom med kvalifisert personale
- Leketerapi
- Sykehusskole
- Spesialisert avlastningstilbud (eks Solhagan på OUS)
- Musikkterapi
- Barnekreftforeningen (www.barnekreftforeningen.no)
2CL-psykiatri står for Consultation-Liaison-psychiatry, et felt som ofte deles i barne-CL og voksen-CL. Innenfor psykisk helse – barn- og unge (tidl. bup) betyr dette å jobbe konsultativt i pediatrien.
3BUP står for Barne- og ungdomspsykiatrisk poliklinikk. Forkortelsen brukes fortsatt, selv om fagfeltet nå heter «Psykisk helse – barn og ungdom».
4DPS står for distriktspsykiatrisk senter, og er et spesialisthelsetjenestetilbud innen psykisk helse for voksne.
5Familievern er nå, sammen med barnevern, organisert under Barne-, ungdoms- og familieetaten. Se www.bufetat.no/familie.
6Statped består av 15 statlig eide kompetansesentre og 25 andre spesialpedagogiske enheter. Disse yter spesialpedagogisk veiledning og støtte til kommuner og fylkeskommuner. Se www.statped.no
7Pedagogisk-psykologisk tjeneste (PPT) er kommunens rådgivende og sakkyndige instans i forhold til spørsmål som omhandler barn, ungdom og voksne som opplever å ha en vanskelig opplærings- eller oppvekstsituasjon.
Rettigheter/støtteordninger
Sist faglig oppdatert: 26.05.2020
Rettigheter/støtteordninger
Med barn menes personer under 18 år. Med foreldre menes også fosterforeldre og foresatte som opptrer i foreldres sted.
Barn på sykehus
Barns rettigheter i sykehus er lovfestet i spesialisthelsetjenesteloven § 3-6 og Forskrift om barns opphold i helseinstitusjon. www.lovdata.no
Samvær med foreldre
Barn har rett til å ha minst en av foreldrene hos seg under innleggelsen. Ved alvorlig eller livstruende sykdom skal begge foreldre kunne få være hos barnet. En av foreldrene som er hos barnet, skal få tilbud om overnatting i institusjonen. Ofte er det aktuelt at foreldre og barn sover i samme rom. Personalet skal avklare med foreldrene hvilke oppgaver foreldrene skal utføre mens de er hos barnet. En ledesnor kan være at foreldrene utfører de samme oppgavene som de gjør når barnet er hjemme, for eksempel stell, påkledning og mating. Foreldre og barn skal få fortløpende informasjon om sykdommen.
Det er en stor belastning å ha et alvorlig sykt barn. Derfor skal foreldre som er hos barnet under sykehusoppholdet, få avlastning etter behov. Helseinstitusjonen skal tilby foreldrene kontakt med sosionom, psykolog og/eller annet støttepersonell mens barnet er innlagt.
Reiseutgifter
Reiseutgifter etter gjeldende satser dekkes i følgende tilfeller:
- Ved innleggelse og utskrivning av barnet med nødvendig følge
- Når institusjonen av medisinske grunner innkaller foreldre for å være hos barnet, for informasjon om sykdomsutvikling eller for nødvendig opplæring
- Når barnet har foreldre hos seg under oppholdet, skal helseinstitusjonene etter 14 dagers liggetid dekke en tur/retur reise i uken for en av foreldrene dersom denne har behov for å reise hjem
Aktivisering/undervisning
Barn kan motta besøk utenom vanlig besøkstid. Personalet skal ta hensyn til at barna trenger besøk, og legge opp rutinene deretter. Besøk kan nektes hvis medisinske årsaker tilsier det.
Barn skal aktiviseres og stimuleres så langt deres helsetilstand tillater det.
Barn i skolepliktig alder har rett til undervisning både i grunnskolen og videregående skole.
Oppfølging på hjemstedet
Sykehuset har ansvar for at det blir etablert kontakt med det kommunale hjelpeapparatet når det er behov for det etter utskrivning. Dette skal gjøres i samråd med foreldrene. Aktuelle instanser kan være: helsesøster, fastlege, skole, barnehage, Pedagogisk Psykologisk Tjeneste.
Individuell plan og koordinator
Barn som har behov for langvarige og koordinerte tjenester fra flere hjelpeinstanser, har rett til å få utarbeidet individuell plan. Ved utarbeidelsen av individuell plan skal det oppnevnes en koordinator. Jfr. helse- og omsorgstjenesteloven §§ 7-1 og 7-2, spesialisthelsetjenesteloven §§ 2-5 og 2-5 a, pasient- og brukerrettighetsloven § 2-5 og forskrift om habilitering og rehabilitering, individuell plan og koordinator.
Kommunene skal tilby koordinator for pasienter med behov for langvarige og koordinerte tjenester, også når pasienten ikke ønsker individuell plan. Det samme gjelder i spesialisthelsetjenesten. Tilbud om koordinator i spesialisthelsetjenesten skal dessuten gis til pasienter med behov for komplekse tjenester. Koordinatoren skal sørge for nødvendig oppfølging av den enkelte pasienten og sikre samordning av tjenesttilbudet og fremdrift i arbeidet med individuell plan. Koordinator i spesialisthelsetjenesten skal sørge for koordinering av tjenestene i forbindelse med sykehusinnleggelse.
Dersom pasienten har behov for tjenester både fra helse- og omsorgstjenesten i kommunen og fra spesialisthelsetjenesten, er det kommunen som har ansvar for at det blir utarbeidet individuell plan. Behov for individuell plan meldes da til koordinerende enhet i kommunen. Helseforetaket skal medvirke til kommunens arbeid med planen.
Minoritetsperspekitivet
For å sikre at alle pasienter tilbys likeverdig behandling, trenger enkelte grupper tilrettelagt oppfølging for å forstå og gjøre seg forstått. Dette kan gi utfordringer i kommunikasjonen mellom pasient, pårørende og helsepersonell. Det kan dreie seg om språkproblemer, ulik sykdomsforståelse og kulturelle og religiøse forskjeller. Kultursensitiv håndtering av forskjeller er nødvendig for å skape tillit og sikre innsikt i ulik sykdomsforståelse.
Lov om pasient- og brukerrettigheter (kap. 3) pålegger helsepersonell en plikt til å gi nødvendig infomasjon. Pasienten skal ha den informasjonen som er nødvendig for å få innsikt i helsetilstanden og i innholdet av helsehjelpen. I de tilfellene hvor pasienten er fremmedspråklig, må behov for tilrettelegging via tolk vurderes.
Foreldres inntekter
Følgende ytelser kan være aktuelle:
Pleiepenger
(folketrygdloven § 9-10)
Den som har omsorgen for et barn under 18 år, har rett til pleiepenger når barnet på grunn av sykdom, skade eller lyte har behov for kontinuerlig tilsyn og pleie, og medlemmet derfor må være borte fra arbeidet. Pleiepenger utbetales etter de samme reglene som ved egen sykdom.
Inntektsgivende arbeid ligger til grunn for ytelsen. Dersom mor eller far mottar foreldrepenger for det syke barnet, skal det søkes om pleiepenger.
Det er en forutsetning at foreldrene av hensyn til barnet må oppholde seg i helseinstitusjonen eller være hjemme fordi barnet trenger kontinuerlig tilsyn og pleie. Også andre enn barnets foreldre kan få pleiepenger dersom det er nødvendig av hensyn til barnet.
Regelverket for pleiepenger ble omgjort i oktober 2017. Det var tidligere et skarpt skille mellom svært alvorlig/ livstruende tilstand og andre tilstander. Dette skillet er opphevet, og det er kontinuerlige pleie- og tilsynsbehovet som legges til grunn.
Når barn er innlagt på sykehus, er det tilstrekkelig at legen dokumenterer at det er behov for at begge foreldre/omsorgspersoner er til stede samtidig. Søkes det om pleiepenger i hjemmet, må legen dokumentere at barnet har et så omfattende pleiebehov at det er nødvendig med mer enn en omsorgsperson med 100 % pleiepenger. Hvis nødvendig kan andre enn foreldre få pleiepenger. Pleiepenger ytes så lenge det er nødvendig i forhold til barnets behov for ekstra tilsyn og pleie.
Graderte pleiepenger
(folketrygdloven § 9-11)
Det kan ytes graderte pleiepenger når barnet trenger kontinuerlig tilsyn og pleie, men hvor det er etablert tilsyns- eller avlastningsordninger deler av dagen eller noen dager i uken. Ytelsen kan graderes ned til 20 % for en eller begge foreldre. Ytes så lenge det er nødvendig.
Opplæringspenger
(folketrygdloven § 9-14)
Foreldre får opplæringspenger hvis de gjennomgår nødvendig opplæring ved en godkjent helseinstitusjon eller et foreldrekurs. Montebello-Senteret arrangerer kurs i samarbeid med Støtteforeningen for Kreftsyke barn (SKB).
Opplæringspenger utbetales etter de samme reglene som ved egen sykdom. Det er legen som må dokumentere behovet for både pleiepenger og opplæringspenger. NAV utbetaler begge ytelsene, evt. refunderer til arbeidsgiver dersom foreldrene arbeider på et sted som har refusjonsavtale med NAV.
Omsorgspenger ved kronisk sykdom
(folketrygdloven §§ 9-5 til 9-9)
Kreftsykdommer hos barn gir rett til 10 ekstra fraværsdager for hver av foreldrene dersom barnets behov for oppfølging medfører økt risiko for fravær fra arbeidet. 20 dager hvis alene med omsorgen. Retten gjelder frem til barnet fyller 18 år.
Hjelpestønad og forhøyet hjelpestønad til barn og unge
(folketrygdloven §§ 6-4 og 6-5)
Hjelpestønad skal gi økonomisk kompensasjon når barnet har behov for særskilt tilsyn og pleie. Barnets ekstra behov for tilsyn og pleie som følge av sykdommen avgjør hvilken sats (4 ulike) som tilstås. NAV har utarbeidet egne retningslinjer for hjelpestønad ved visse typer lidelser, bl.a. barn med kreft. Når diagnosen er stilt og behandling igangsatt, antas kriteriene å være oppfylt. Det konkrete tilsyns- og pleiebehovet må imidlertid beskrives i søknaden, slik at riktig sats kan fastsettes. Ytelsen kan tilstås fra måneden etter diagnosen ble stilt.
Grunnstønad
(folketrygdloven § 6-3)
Grunnstønad skal dekke ekstra utgifter som følge av sykdommen. Disse kan være: økte utgifter til transport, ekstra slitasje på klær og sengetøy grunnet hyppig vask. De ekstra utgiftene må minst tilsvare sats 1 for å bli innvilget, og må sannsynliggjøres og helst dokumenteres. Kvitteringer på ekstra utgifter til ekstra slitasje på klær og sengetøy (innkjøp) for minimum 3 måneder må som hovedregel vedlegges søknaden.
Avlastning/støttekontakt
(helse- og omsorgstjenesteloven §§ 3-2 og 3-6)
Oppholdskommunen skal yte avlastningstiltak til familier som har særlig tyngende omsorgsarbeid. Antall timer bestemmes ut fra en helhetlig og skjønnsmessig vurdering av kommunen.
Omsorgsstønad
(helse- og omsorgstjenesteloven § 3-6)
Ved tyngende omsorgsoppgaver i hjemmet kan foreldre søke kommunen om omsorgsstønad. Det skal ikke tas hensyn til familiens økonomiske forhold utover tilstått hjelpestønad. Det er imidlertid viktig å være klar over at denne ytelsen regnes som inntekt – i den forstand at dersom en av foreldrene har NAV-ytelser som eksempelvis arbeidsavklaringspenger, avkortes NAV-ytelsen tilsvarende. Da bør i så fall den andre forelderen stå som mottaker av omsorgsstønaden, evt. (dersom begge foreldre er mottakere av NAV-ytelsen, eller forelderen er alene om omsorgen), at NAV gjøres kjent med at omsorgsstønad er innvilget.
Studenter
Ved sykdom hos barn under 12 år som hindrer forelderen i å følge undervisningen, kan Lånekassen omgjøre lån til sykestipend (www.lanekassen.no)
Overgangsstønad
(folketrygdloven § 15-5 og § 15-9)
Tidsbegrenset stønad til enslig mor eller far som er midlertidig ute av stand til å forsørge seg ved eget arbeid eller er under utdanning. Dersom barnet er særlig tilsynskrevende på grunn av alvorlig sykdom, kan ytelsen forlenges.
Vaksinasjoner ved barnekreft
Sist faglig oppdatert: 26.05.2020
Flere studier har vist at vaksinasjonsbeskyttelsen er dårlig etter gjennomgått kreftbehandling. Rekonstitusjon av immunsystemet tar tid, avhengig av diverse faktorer som behandlingsintensitet og alder, og kan variere fra barn til barn. Vaksinasjon kan foregå i vedlikeholdsfasen av behandlingen, men gir ofte dårlig beskyttelse. Alle barn som har gjennomgått kreftbehandling anbefales oppvaksinering etter avsluttet behandling; prinsipielt så tidlig som mulig, men under forutsetningen at det har tilkommet tilstrekkelig immunologisk rekonstitusjon etter kreftbehandlingen for å kunne utløse en vaksinerespons (Abzug, 2009; Australian Technical Advisory Group on Immunisation (ATAGI), 2018; Esposito, Cecinati, Brescia, & Principi, 2010; Reilly et al., 2010; Rubin et al., 2014; Saghafian-Hedengren, Soderstrom, Sverremark-Ekstrom, & Nilsson, 2018; van Tilburg et al., 2011; Wiegering et al., 2014).
Prinsipper ved vaksinasjon hos immunsupprimerte
Ved vaksinering hos immunsupprimerte, herunder barn under pågående/nylig avsluttet kreftbehandling, må man vurdere sikkerhet og effekt av vaksinering.
Vaksiner med levende, svekkede mikrober bør unngås under pågående behandling og ved residivmistanke da vaksinevirus kan gi sykdom hos immunsupprimerte.
Alle inaktiverte vaksiner kan gis til immunsupprimerte barn. Redusert respons av vaksinering er ikke grunnlag for å unnlate vaksinering, og delvis vaksinasjonsbeskyttelse er bedre enn ingen beskyttelse. Unntak er vaksinasjon under behandling med Rituximab, hvor behandlingen medfører manglende vaksineeffekt i 6 måneder etter siste dose.
Det anbefales samråd med infeksjonsspesialist eller vaksinelege på Folkehelseinstituttet (telefonnr. 21077000) ved usikkerhet om risiko, uklarheter omkring grunnbeskyttelsen, reisevaksiner og andre spesialtilfeller.
Barn som starter kreftbehandling i første leveår eller andre uvaksinerte barn
Barn som blir syke i første leveår eller av andre årsaker ikke er fullvaksinerte før kreftdiagnose, anbefales å starte barnevaksinasjonsprogrammet på nytt etter avsluttet kreftbehandling (tabell 1). Det er mulig å gi inaktiverte vaksiner under pågående behandling i perioder med lav behandlingsintensitet (f. eks vedlikeholdsfasen), men i denne fasen kan vaksinering gi lav grad av beskyttelse.
Generelle råd ved vaksinering av barn med kreft
Nøytropeni: Unngå vaksinering ved nøytrofile granulocytter < 0,5 x 109/L fordi evt bivirkning i form av feber etter vaksine vil medføre innleggelse og eventuell behandling med antibiotika.
Rituximab: unngå vaksinering med levende, svekkede vaksiner i minimum 6 måneder etter behandling. Avvente inaktiverte vaksiner i 6 måneder, da det ikke kan påregnes tilstrekkelig respons på vaksinen(e).
Steroidbehandling: Levende, svekkede vaksiner er kontraindiserte til barn som får høye steroiddoser (≥ 2 mg/kg/døgn). Høye steroiddoser kan gi dårligere vaksinerespons mot inaktiverte vaksiner, men kunnskapen er begrenset. Det antas at pasienter som bruker systemiske steroider i moderate doser (<2 mg/kg/d), vanligvis responderer tilfredsstillende på inaktiverte vaksiner. Ved langvarig behandling med moderate doser steroider, bør det vurderes pause i steroidbehandlingen dersom det er behov for levende, svekkede vaksiner.
Immunglobulin: Avvente levende, svekkede vaksiner de første 3 måneder etter tilførsel av immunglobulin, da vaksineantigenet kan inaktiveres av antistoffer i det tilførte immunglobulinet/blodproduktet og vaksinen da kan ha redusert effekt.
Nærkontakter: det anbefales vaksinasjon av husstandsmedlemmer og andre nærkontakter for å minske smitteeksponering til den syke. Dette gjelder også ved bruk av vaksiner med levende, svekkede mikrober. Mest aktuelle vaksiner: influensa, varicella, oppdatert beskyttelse mot difteri-stivkrampe-kikhoste-polio.
Antistoffmåling: Ved diagnose blir det tatt varicellaantistoffer før behandlingsstart og før det gis blodprodukter. Pasienter seronegative for varicella skal ha profylakse ved eksponering under kreftbehandling og vaksinasjon etter behandling. For andre vaksineantigener er det ikke nødvendig å måle antistoffer før oppstart av kreftbehandling eller for å vurdere behov for vaksinasjon etter avsluttet behandling.
Posteksponering: Ved eksponering for varicella se avsnitt nedenfor. Ved eksponering for meslinger, kan det gis normaltimmungloblulin,se nettsidene til Folkehelseinstituttet: https://www.fhi.no/nettpub/vaksinasjonsveilederen-for-helsepersonell.
Ansvar for vaksinasjon: Den behandlende enheten har kunnskapen om pasientens immunstatus og bør derfra ta det overordnede ansvaret for vaksinasjonsanbefalinger. Selve vaksinasjonen kan delegeres til andre enheter etter avtale (fastlege, helsestasjon, vaksinasjonskontor).
Videre vaksinasjon: etter fullført oppvaksinering etter kreftbehandling, kan barna generelt tilbys de neste vaksinene i barnevaksinasjonsprogrammet ved samme alder som sitt årskull.
Vaksinasjon av barn etter avsluttet kreftbehandling
Antistoffnivåene reduseres under behandlingen, og mange har antistoffmengder under beskyttende nivå for ett eller flere antigen. Det anbefales derfor å gi boosterdoser etter avsluttet kreftbehandling til alle. Antistoffnivået behøver ikke måles for å vurdere behov for revaksinering. Hos tidligere ufullstendig vaksinerte barn (kreftsykdom i første leveår eller manglende vaksinasjon av andre årsaker) anbefales det å starte vaksinasjon på nytt.
Tabell 1: Anbefalt vaksinering etter avsluttet kreftbehandling
OBS: barn som ikke har fullført barnevaksinasjonsprogrammet første leveår eller av andre grunner ikke er fullvaksinerte, anbefales å starte vaksinasjonsprogrammet på nytt – se tabell 2 lengre ned og tekst
| Vaksine | Nr i teksten nedenfor | Lavintensiv cytostatikabehandling (eks ALL-SR+IR, Hodgkin TG1, B‑NHL R1+R2 (uten Rituximab), Wilms st 1–2 AV-beh, RMS LR, NB LR, LGG) | Høyintensiv cytostatikabehandling (eks ALL-HR, AML, T-NHL, B-NHL R3 + R4 (+ etter Rituximab) , Hodgkin TG2–3, Wilms AVD/HR-beh, RMS-SR/HR, ES, OS, NB-HR, Medulloblastom) | Kommentar |
| DTP-polio-Hib-HepB | 1 | 3 (6 md ved bruk av Rituximab) | 6 + 8 | Barn som får kreft i første leveår: egne anbefalinger |
| Pneumokokk PKV13 | 2 | 3 (6 md ved bruk av Rituximab) | 6 | Barn som får kreft i første leveår: egne anbefalinger |
| Pneumokokk PPV23 | 2 | 1 dose minst 8 uker etter PKV13 vurderes for barn > 2 år med særlig risiko for alvorlig pneumokokksykdom | ||
| MMR | 3 | 3–6 (6 md ved bruk av Rituximab) | 6–12
| Total lymfocytter > 1,2 x 109/L Ved bruk av Rituximab skal levende vaksine utsettes til minst 6 måneder etter avsluttet behandling. 2 doser hvis eldre enn 6. klasse (når 2. dose gis i barnevaksinasjonsprogr.) |
| HPV, jenter (alle) og gutter født 2006 eller senere Eldre gutter: se tekst | 4 | Vaksinering kan foregå når barnets årskull skal tilbys vaksinen i barnevaksinasjonsprogrammet (7. klasse). Boosterdose anbefales til de som har gjennomgått kreftbehandling etter gjennomført HPV-vaksinasjon, se tekst. Vaksinering kan starte 6 måneder etter avsluttet behandling. | ||
| Influensavaksine (inaktivert type) alder > 6 mnd | 5 | Hver sesong under pågående kreftbehandling og inntil 6 mnd etter avsluttet behandling. Ved førstegangsvaksinering hos barn under 9 år skal barnet ha 2 doser influensavaksine med 4 ukers intervall. | ||
| Varicellavaksine alder > 9 mnd Total lymfocytter > 1,2 x 109/L)*** | 6 | 6+8 | 6+8 | Total lymfocytter > 1,2 x 109/L) Tilbys kun barn som ikke har hatt vannkopper |
Mer om de enkelte vaksinene:
Seksvalent vaksine mot Difteri, tetanus, pertussis, polio, Hib, og Hepatitt B (DTP-IPV-Hib-HepB)
Alle barn tilbys nå en seksvalent kombinasjonsvaksine mot difteri, tetanus, pertussis, polio, Hib og hepatitt B (DTP-polio-Hib-HepB) i barnevaksinasjonsprogrammet. Den seksvalente vaksinen kan også brukes til oppvaksinering av barn etter kreftbehandling, selv om disse ikke har fått vaksine mot hepatitt B tidligere i barnevaksinasjonsprogrammet. Alternativt gis DTP-IPV (firevalent fulldosevaksine, Tetravac®) og monokomponent Hib-vaksine (Act-Hib®). Ingen av disse vaksinene inneholder levende komponenter.
Anbefalinger for revaksinasjon:
Barn som har påbegynt kreftbehandling i løpet av første leveår og ikke har fått alle doser første leveår, bør starte vaksinasjon på nytt. Oppstart senest 3–6 måneder etter avsluttet behandling (se tabell 1). Det er mulig å gi inaktiverte vaksiner under pågående behandling i perioder med lav behandlingsintensitet (f. eks vedlikeholdsfasen), men i denne fasen kan vaksinering gi lav grad av beskyttelse.
Barn som har fått minst 3 doser før diagnose, anbefales 1–2 boosterdoser 3–6 måneder etter avsluttet behandling avhengig av hvor intensiv behandlingen har vært (se tabell 1).
Ved bruk av Rituximab skal vaksinasjon utsettes til det har gått minst 6 måneder fra siste behandling.
Etter høyintensiv kjemoterapi, er én dose kombinasjonsvaksine vanligvis ikke tilstrekkelig for å oppnå beskyttelse. Det er usikkert om det er behov for ny, komplett grunnimmunisering, eller om det er tilstrekkelig med 2 doser. Ved høyintensiv behandling anbefales minst 2 doser fra 6 måneder etter avsluttet behandling. Neste dose gis 2 måneder senere.
I risikosituasjoner (sårskader, utlandsreise, utbrudd av kikhoste eller difteri) gis en boosterdose uansett tidspunkt.
Pneumokokkvaksine
https://www.fhi.no/nettpub/vaksinasjonsveilederen-for-helsepersonell(Abzug, 2009; Australian Technical Advisory Group on Immunisation (ATAGI), 2018)
Det finnes to vaksiner mot pneumokksykdom. Begge inneholder kun inaktiverte komponenter.
Pneumokokk konjugatvaksine (PKV13, mot 13 pneumokokkserotyper) inngår i den norske barnevaksinasjonsprogrammet. Vaksinen ble introdusert (da som PKV7) i juli 2006, og barn før dette har ikke fått tilbud om denne vaksinen i spedbarnsalder. PKV13 gir immunologisk hukommelse.
Pneumokokk polysakkaridvaksine (PPV23, mot 23 pneumokokkserotyper) gir ikke immunrespons hos barn under 2 år. Vaksinen gir heller ikke immunologisk hukommelse. Vaksinen anbefales til pasienter > 2 år med økt risiko for invasiv pneumokokksykdom grunnet for eksempel immunsupprimerende behandling. Vaksinen dekker for flere serotyper enn PKV13. Det er imidlertid få tilfeller av invasiv pneumokokksykdom hos barn som skyldes serotyper som dekkes i PPV23 men ikke i PKV13, altså vil det for de fleste barn ikke være nødvendig med PPV23 i tillegg til PKV13.
Ved diagnosetidspunkt (Bate et al., 2019):
Uvaksinerte barn anbefales å få 1 dose PKV13 ved diagnosetidspunktet. Som uvaksinert regnes spedbarn som ikke har rukket å få første dose PKV13 eller innvandrerbarn fra land hvor ikke PKV tilbys. Etter avsluttet kreftbehandling skal disse barna oppvaksineres ihht anbefaling for alder (egne anbefalinger hvis <1 år ved diagnose) eller behandlingsintensitet (tabell 1).
Anbefaling revaksinasjon etter avsluttet kreftbehandling:
Barn som har påbegynt kreftbehandling i løpet av første leveår og ikke har fått alle 3 doser med PKV13, anbefales å starte vaksinasjon på nytt etter avsluttet behandling med oppstart 3–6 måneder etter behandlingsslutt (se tabell 1). Barnet kan vaksineres i passende faser så snart de er i remisjon (f.eks under vedlikeholdsbehandling), men nyere studier har vist at dette ikke gir særlig god beskyttelse.
Barn som har fått sine tre doser med PKV13, tilbys en boosterdose med PKV13 3–6 måneder etter avsluttet behandling (se tabell 1).
Ved bruk av Rituximab skal vaksinasjon utsettes til det har gått minst 6 måneder fra siste behandling
For barn > 2 år med spesielt høy risiko for pneumokokkinfeksjon eller tilleggssykdommer, kan det vurderes om det er behov for 1 dose PPV23 etter avsluttet kreftbehandling. Dette kan for eksempel gjelde barn som har begrenset lungefunksjon etter gjennomgått behandling, eller som står på immunsupprimerende behandling. Til disse skal alltid PKV13 gis først, og PPV23 gis tidligst 8 uker senere: https://www.fhi.no/publ/2015/anbefalinger-for-bruk-av-pneumokokk/
Meslinger-kusma-røde hunder (MMR)
Vaksinen inneholder levende svekket virus og skal ikke gis til pasienter under kreftbehandling.
Alle barn som har avsluttet kreftbehandling, anbefales minst en rutinemessig boosterdose etter avsluttet behandling. Denne dosen kan gis 3–12 måneder etter avsluttet behandling avhengig av behandlingsintensitet (se tabell 1). Jo mer intensiv behandling, jo lengre tid bør gå etter avsluttet behandling før vaksine gis.
Ved bruk av Rituximab skal vaksinasjon utsettes til det har gått minst 6 måneder fra siste behandling.
Videre kan barn følge det allmenne vaksinasjonsprogrammet med en booster ved 11–12 års alder. For barn eldre enn dette, må det påses at det gis 2 doser MMR-vaksine med minst 3 måneders intervall.
Vaksine mot humant papillomavirus (HPV)
Vaksinen inneholder kun inaktiverte komponenter. Vaksinen tilbys jenter (fra årskull 1997) og gutter (fra årskull 2006) i 7. klasse. Personer som får immunsupprimerende behandling har økt risiko for HPV-relatert kreft, og pasienter som har gjennomgått behandling for kreft har økt risiko for utvikling av sekundær kreft. På denne bakgrunn anbefales:
Jenter og gutter før 7.klasse og som ikke har fått HPV-vaksine før påbegynt kreftbehandling, kan få vaksinen fra 6 måneder etter avsluttet behandling eller ved samme alder som det ellers tilbys i barnevaksinasjonsprogrammet (7. klasse). Barn som nylig har gjennomgått kreftbehandling anbefales 3 doser (minst 1 måned mellom de første to doser og minst 5 måneder mellom de siste to doser) da det mangler dokumentasjon på langtidsbeskyttelse av 2 dose-regime ved immunsuppresjon.
Jenter (alle) og gutter (født 2006 eller senere) som har fått en dose HPV-vaksine før påbegynt kreftbehandling, anbefales 3 doser (minst 1 måned mellom de første to doser og minst 5 måneder mellom de siste to doser), fra 6 måneder etter avsluttet kreftbehandling, da det mangler dokumentasjon på langtidsbeskyttelse av 2 dose-regime ved immunsuppresjon/nylig avsluttet kreftbehandling.
Jenter og gutter som har fått to eller tre doser HPV-vaksine før påbegynt kreftbehandling, bør tilbys en dose HPV-vaksine fra 6 måneder etter avsluttet kreftbehandling.
Gutter som har gjennomgått kreftbehandling og som ikke har tilbud om HPV-vaksine i barnevaksinasjonsprogrammet (født 2005 eller tidligere), bør også vurdere å la seg vaksinere mot HPV grunnet risiko for senere kreft. Vaksinen må da bekostes av barnets foreldre/foresatte. Vaksinasjon kan påbegynnes 6 måneder etter kreftbehandling.
Influensavaksine
Influensavirus er et vanlig luftveisvirus som opptrer epidemisk i vinterhalvåret, og kreftsyke barn har løpende risiko for alvorlig sykdom, opphold i kreftbehandlingen, hospitalisering og antibiotikabehandling ved infeksjon. Immunsupprimerte barn har også større risiko for virusutskillelse over lang tid og bidrar til smittespredning.
Det finnes to typer influensavaksine; inaktivert injeksjonsvaksine (trivalent inaktivert vaksine) og levende, svekket influensavirus («live attenuated influenza vaccine» – LAIV) som gis nasalt. Kun den inaktivert vaksinen er anbefalt barn under pågående kreftbehandling og til nærkontakter til pasienter som er svært immunsvekket. Barn med ALL har bedre respons på influensavaksine gitt i induksjonsfase enn i vedlikeholdsfase, og kan derfor vaksineres uavhengig av behandlingsfase. Barn med solide svulster har om lag samme respons på vaksine uavhengig av hvor lenge de hadde fått behandling.
Barn under pågående kreftbehandling anbefales influensavaksine hver sesong inntil 6 måneder etter avsluttet behandling før ny influensasesong. Det finnes flere ulike injeksjonsvaksiner, to av dem kan gis fra alder 6 måneder og alle kan gis fra 3-årsalder. Ved førstegangs vaksinasjon hos barn under 9 år skal det gis 2 doser med 4 ukers intervall.
Vaksinering av nærkontakter og helsepersonell anbefales for å minske pasientens eksponering for influensavirus.
Influensavaksinasjonen gir bare delvis beskyttelse og barnet kan derfor fortsatt få influensa.
Vannkoppevaksine og post-eksponeringsprofylakse
Vaksinen inneholder levende, svekket virus og skal ikke gis til pasienter under (intensiv) kreftbehandling.
Anbefalinger vedrørende bruk av varicellavaksine til barn med kreft og etter kreftbehandling er sprikende. Dødelighet av varicellainfeksjon blant barn med kreft synes å være lav ved retrospektiv gjennomgang av barn i forskjellige ALL-protokoller (Caniza et al., 2012). På den andre siden har det vært rapportert et dødsfall hos et barn med ALL i vedlikeholdsfase etter varicellavaksinasjon der infeksjon med vaksinevirus var årsaken (Schrauder et al., 2007). I Danmark blir seronegative barn (ved diagnose) med ALL blitt anbefalt varicellavaksinasjon under pågående vedlikeholdsbehandling, med gode resultater (Smedegaard et al., 2016). Den norske anbefalingen er forsiktig sammenlignet med den danske, men i tråd med andre nasjoners anbefalinger. Dersom det skulle være situasjoner der det er ønske eller behov for å vaksinere barn under pågående kreftbehandling, anbefales det å følge den danske referanseartikkelen. Folkehelseinstituttet anbefaler generelt å vaksinere når total lymfocytter er > 1,2 x 109/L.
Varicellavaksine
Nærkontakter: Mottagelige (=seronegative) søsken (alder > 9 måneder), foreldre og helsepersonell kan vaksineres om man vil minske risikoen for eksponering i husstanden. Cave: graviditet! Etter vaksinasjon er det ugunstig med besøk på sykehuset i de neste 3 uker fordi eventuelt vaksineutslett forårsaker usikkerhet om den vaksinerte er smittet med vannkopper samtidig. Derimot behøver man ikke skille søsken fra det kreftsyke barnet, med mindre vaksinert søsken utvikler vaksineutslett hvor en er usikker på om det er vannkopper samtidig. Gi da posteksponeringsprofylakse til den kreftsyke (Marin, Leung, & Gershon, 2019).
Pasienter: Primært seronegative pasienter kan få Varicellavaksine 6+8 måneder etter avsluttet behandling.
Etter to doser varicellavaksine vil ca 95 % av immunfriske barn være beskyttet mot VZ-virus, og vaksinering er særskilt viktig for den gruppen som senere får kreftresidiv.
Primært seropositive behøver ikke vaksineres selv om de har lave antistoffnivå etter avsluttet behandling. Cellulært immunforsvar bevares ofte.
Posteksponeringsprofylakse
Primært antistoffnegative pasienter under og de første 3 månedene etter kreftbehandling skal ha acyclovir (eller Valtrex) i varicelladose i 14 dager fra dag 5–7 etter eksponering (se også kapittel 3.22 i Generell veileder i pediatri: https://www.helsebiblioteket.no/pediatriveiledere?menuitemkeylev1=5962&menuitemkeylev2=5965&key=247451.
Primært seropositive pasienter bør få posteksponeringsprofylakse ved eksponering i intensive behandlingsfaser (se kap. 3.22 i Generell veileder i pediatri) Generell veileder for pediatri, eller ved kraftig eksponering (f.eks dersom søsken med varicella).
Det norske barnevaksinasjonsprogrammet per 2019
Etter fullført oppvaksinering etter gjennomført kreftbehandling, kan de generelt tilbys de neste vaksinene i barnevaksinasjonsprogrammet ved samme alder som sitt årskull.
Tabell 2: Det norske barnevaksinasjonsprogrammet f.o.m. august 2018
Vaksiner i kursiv består av levende, svekkede mikrober
| Vaksine mot | Gis ved alder | Relevante opplysninger |
|---|---|---|
| Rotavirus | 6+12 uker | Begge doser skal være gitt før senest alder 16 uker. Det er ikke aktuelt å tilby oppvaksinering etter fullført kreftbehandling, uansett alder |
| DTP-polio-Hib-HepB (grunnvaksinering, fulldose) | 3+5+12 måneder | Hepatitt B til alle barn født fra 01/11/2016, før det til barn med foreldre fra endemiske land |
| Pneumokokk konjugat vaksine mot 13 serotyper (PKV13) | 3+5+12 måneder | PKV7 (mot 7 serotyper) innført for barn født fra 2006 PKV13 fra 2011 |
| MMR | 15 måneder og 6. klasse |
|
| DTP-polio (1. boosterdose, fulldose) | 2. klasse (noen steder 1. klasse) |
|
| HPV | 7. klasse, 2 doser med minst 6‑måneders intervall | Jenter tilbud fra årskull 1997, gutter fra årskull 2006 |
| DTP-polio (2. boosterdose, lavdose) | 10. klasse |
|
| BCG (kun til risikogrupper) | 6 uker | Sjelden aktuelt hos større barn etter avsluttet kreftbehandling. Ikke til immunsupprimerte. |
Vaksinasjoner etter allogen stamcelletransplantasjon til barn
Sist faglig oppdatert: 26.05.2020
(Australian Technical Advisory Group on Immunisation (ATAGI), 2018; Carreras, Dufour, Mohty, & Kröger, 2019; Ljungman et al., 2009; Rubin et al., 2014)
Pasienter som har gjennomgått stamcelletransplantasjon (allogen eller autolog) har nedsatt infeksjonsforsvar i lang tid etter transplantasjonen. De fleste pasienter mister sin immunitet mot infeksjoner som de har gjennomgått eller er blitt vaksinert mot tidligere i livet.
Pasienter som har gjennomgått stamcelletransplantasjon skal rutinemessig tilbys revaksinasjon. Grunnlaget for disse retningslinjene er basert på internasjonale anbefalinger tilpasset norske forhold. Det foreligger ingen publiserte data som tilsier at pasienter som transplanteres for ulike diagnoser med ulik kondisjonering, giver eller stamcellekilde (allogen eller autolog) behøver særlig tilpasset vaksinasjonsprogram.
Ved utskrivning etter transplantasjon eller senest ved 3-månederskontroll ved OUS/Rikshospitalet får pasient et informasjonsskriv vedlagt vaksinasjonsskjema og utdypende informasjon om de enkelte vaksinene. Anbefaling om vaksinasjon er behandlende leges ansvar, men vaksinasjonen kan gjennomføres av den lokale helsestasjon eller pasientens fastlege etter avtale. Gitte vaksiner skal dokumenteres i nasjonalt vaksinasjonsregister SYSVAK og i vaksinasjonsskjemaet som returneres til transplantasjonskoordinator, Barne- og ungdomsklinikk, OUS-Rikshospitalet.
Vaksinasjonsanbefalinger
Vaksinasjonsskjemaet er basert på internasjonale anbefalinger, men kan tilpasses individuelt.
Spesielle hensyn hos stamcelletransplanterte:
Pasienter med kronisk GvHD: skal ikke motta levende, svekkede vaksiner men kan ellers vanligvis vaksineres etter samme skjema som andre. Det er ingen grunn til å hevde at pasienter risikerer aktivering av GvHD som følge av vaksinering, men det er sannsynlig at de vil respondere noe dårligere enn pasienter uten GvHD og immunsuppresjon.
Pågående immunsuppressiv behandling: immunsuppressiv behandling være seponert minst 3 måneder før vaksinasjon med levende, svekkede vaksiner. Ved bruk av Rituximab bør all vaksinasjon utsettes til 6 måneder etter siste dose Rituximab.
IVIG: Pasienter som fortsatt trenger IVIG ved oppstart av revaksineringen 3 måneder etter transplantasjonen (første pneumokokkvaksine), kan starte revaksineringen i henhold til vaksineskjema, men bør kontrollere vaksineresponsen (pneumokokk vaksinetiter, analyseres på FHI) 3 måneder etter at den siste IVIG dosen ble gitt. Se også avsnitt om pneumokokkvaksinen. Det anbefales å vente minst 3 måneder etter en IVIG infusjon før man gir vaksiner med levende, svekkede mikrober.
Tabell 3: Anbefalt vaksinasjon etter stamcelletransplantasjon (autolog eller allogen)
NB: Ved bruk av Rituximab bør all vaksinasjon utsettes til 6 måneder etter siste dose Rituximab
| Vaksine | Gjelder | Tidspunkt etter transplantasjon | ||||||
|---|---|---|---|---|---|---|---|---|
| Pneumokokk Prevenar 13 (PKV13) | Gis til alle | 3 mnd | 4 mnd | 5 mnd | 12 mnd: Pasienter med kronisk GVHD* | |||
| Pneumokokk Pneumovax (PPV23) | Gis til pasienter uten GvHD | Gis 12 mnd etter transplantasjon istedenfor den 4. dosen Prevenar13 (PKV 13), men kun til pasienter > 2år uten kronisk GvHD og uten pågående immunsuppresjon | ||||||
| Difteri, tetanus, kikhoste, inaktivert polio, Haemophilus influenza type b, hepatitt B Kombinasjonsvaksine: Infanrix-hexa eller Hexyon | Gis til alle | 6 mnd | 7 mnd | 12 mnd | ||||
| Influensa (inaktivert vaksine) | Gis til alle | Gis i forbindelse med influensasesongen når vaksine er tilgjengelig, tidligst 3 mnd etter transplantasjon. 2 doser med 4 ukers intervall til barn under 9 år | ||||||
| HPV-vaksine | Gis til alle jenter og gutter SCTx etter 7. klasse (gutter født 2006 eller senere) | Kan gis fra 6 mndr | 7 mnd (dose 2 gis 1 mnd etter dose 1) | 12 mnd (dose 3 gis 5 mnd etter dose 2) | ||||
| Meslinger, røde hunder og kusma (levende, svekket vaksine) | Individuelt | Etter 24 mndr hos pasienter som ikke har kronisk GvHD* eller mottar immunhemmende behandling
| ||||||
| Varicella zoster (levende, svekket vaksine) | Individuelt | Etter 24 mndr som ikke har kronisk GvHD* eller mottar immunhemmende behandling
| ||||||
| Meningokokk | Individuelt | Anbefales på lik linje med ungdom generelt, men 2 doser med 8 ukers intervall | ||||||
Omtale av de enkelte vaksiner som gis etter stamcelletransplantasjon
Vaksiner i barnevaksinasjonsprogrammet:
Kombinasjonsvaksine mot difteri, tetanus, kikhoste, polio, Hib og hepatitt B
3 doser gis fra 6 måneder etter allogen stamcelletransplantasjon. Kombinasjonsvaksine med fulldose antigen mot difteri, tetanus, kikhoste, inaktivert poliovirus, Haemophilus influenzae type b og hepatitt B skal benyttes. Vaksinen har kun inaktiverte komponenter.
Meslinger, røde hunder og kusma
Vaksinen består av levende, svekket virus. Vaksinering anbefales tidligst 2 år etter gjennomgått stamcelletransplantasjon, men ikke til pasienter med kronisk GvHD og/eller som mottar immunsuppresjon eller har mottatt slik behandling siste 3 måneder. Grunnvaksinering består av 2 vaksinedoser med minst 3 måneders intervall, men det kan med fordel gå lengre tid (for eksempel 1 år, eller hos yngre barn avvente til vanlig vaksinasjonstidspunkt for andre dose i Barnevaksinasjonsprogrammet).
Vaksine mot humant papillomavirus (HPV)
Tilbys gjennom barnevaksinasjonsprogrammet til barn i 7.klasse (til jenter fra årskull 1997 og gutter fra årskull 2006). Barn som har gjennomgått stamcelletransplantasjon kan få HPV-vaksine i barnevaksinasjonsprogrammet når de får tilbud gjennom vaksinasjonsprogrammet i 7.klasse. For barn som transplanteres etter 7.klasse, kan revaksinering starte når det har gått minimum 6 måneder etter transplantasjon. Det er ikke tilstrekkelig erfaring med langtidsbeskyttelse ved 2-doseregime hos immunsupprimerte (inkludert stamcelletransplanterte) og det anbefales derfor 3 doser til disse pasientene (minst 1 måned mellom de første to dosene, og minst 5 måneder mellom de siste to dosene). Vaksinen består av ikke-levende komponenter
Rotavirusvaksine
Vaksinen består av levende, svekket virus, og inngår i det norske Barnevaksinasjonsprogrammet. Vaksinen er kun godkjent for bruk til barn i første levehalvår, og derfor ikke aktuell til bruk hos barn som har gjennomgått stamcelletransplantasjon.
Tuberkulose
BCG vaksine består av levende, svekket bakterie og er kontraindisert hos stamcelletransplanterte.
Pneumokokker
Pneumokokker kan gi alvorlige og potensielt dødelige infeksjoner som pneumoni, sepsis og meningitt. Vaksinasjon mot pneumokokker er derfor svært viktig å få gjennomført hos de stamcelletransplanterte. Studier har vist at det er en fordel å starte vaksinasjon tidlig hos disse pasientene, selv om vaksineresponsen er svakere (Cordonnier et al., 2009).
Pneumokokk-konjugatvaksinen (PKV13, Prevenar 13) inngår i barnevaksinsjonsprogrammet og gir en tidligere vaksinasjonsrespons og immunologisk hukommelse i motsetning til polysakkarid varianten (PPV23, Pneumovax).
Anbefalinger for pneumokokkvaksine:
- Ved 3, 4 og 5 måneder etter transplantasjon: 3 doser PKV13 med 1 måneds mellomrom med oppstart 3 måneder etter transplantasjon (dvs. 3 mnd, 4 mnd og 5 mnd etter transplantasjon) (Cordonnier et al., 2009). Ved bruk av Rituximab bør all vaksinasjon utsettes til 6 måneder etter siste dose Rituximab.
Ved 12 måneder etter transplantasjon, avhengig av GvHD og immunsuppresjon: Pneumovax (PPV23) gir en bredere beskyttelse mot flere pneumokokkserotyper enn Prevenar 13 (23 mot 13 serotyper), men gir dårlig immunsvar de første 12 måneder etter transplantasjon, særlig hos pasienter med kronisk GvHD. Til pasienter uten GvHD og uten pågående immunsuppresjon kan det gis 1 dose Pneumovax 12–14 måneder etter transplantasjon. Hos pasienter med kronisk GvHD eller pågående immunsuppresjon 12 måneder etter transplantasjon, anbefales det å gi en fjerde dose Prevenar13 (minst 6 måneder etter den 3. dosen) i stedet for Pneumovax. Ved forsinket start av PKV13 etter transplantasjon, bør det gå minst 6 måneder fra 3.dose PKV13 til 4.dose med pneumokokkvaksine (enten PKV13 eller PPV23).
IVIG: Pasienter som behandles med intravenøs immunglobulin (IVIG) kan respondere dårligere på vaksiner. For å sikre at det ble utløst en vaksinerespons, kan det måles pneumokokk vaksinetiter (sendes til Folkehelseinstituttet) 3 mnd etter at den siste dosen IVIG ble gitt. Ved suboptimal vaksinerespons må man eventuelt gi supplerende vaksinedoser. Frem til responsen er bekreftet fortsetter pasienten med penicillin. Ved spesielt høy risiko for infeksjoner med kapselkledde bakterier (f.eks. ved metabolsk sykdom eller aspleni) settes pasienten på penicillin profylakse etter transplantasjon og frem til det er gitt minst 3 doser pneumokokkvaksine.
Vaksiner som ikke inngår i barnevaksinasjonsprogrammet
Influensa
Influensa kan gi alvorlige infeksjoner og i tillegg bane vei for bakterielle komplikasjoner. Til pasienter som har gjennomgått stamcelletransplantasjon anbefales det kun å benytte den inaktiverte injeksjonsvaksinen mot influensa. Den levende, svekkede nasale vaksinen er kontraindisert til denne gruppen. Inaktivert vaksine gis i forbindelse med influensasesongen når vaksine er tilgjengelig, som regel i september og oktober, og tidligst 3(–4) måneder etter gjennomgått stamcelletransplantasjon. Ved bruk av Rituximab bør all vaksinasjon utsettes til 6 måneder etter siste dose Rituximab. Til barn < 9 år anbefales 2 doser gitt med 4 ukers intervall. Deretter kan det gis en dose vaksine hvert år de første 2 årene etter transplantasjon, og lengre til pasienter med pågående immunsuppressiv behandling.
Varicella/zoster
Vaksine består av levende, svekket virus. Den må ikke gis til pasienter med kronisk GvHD og/eller som mottar immunsuppresjon eller har mottatt slik behandling siste 3 måneder, og tidligst 2 år etter gjennomgått stamcelletransplantasjon. Vaksinen anbefales til alle barn som har gjennomgått stamcelletransplantasjon. Det skal gis 2 doser med minimum 6 ukers intervall, men det kan med fordel gå lengre tid (for eksempel noen måneder).
Meningokokkvaksine https://www.fhi.no/publ/2014/meningokokksykdom-i-norge-og-anbefa/
Anbefales til pasienter med funksjonell aspleni. Kan tilbys stamcelletransplanterte på lik linje med anbefalinger til befolkningen generelt og i utbruddssituasjoner. Begge typer vaksiner (kombinasjonsvaksine/konjugatvaksine mot meningokokk serogrupper A, C, W135, Y og proteinvaksine mot meningokokk serogruppe B) er inaktiverte/ikke-levende. Vaksinen mot meningokokk serogrupper ACWY har vist seg å være effektiv hos transplanterte, men da behøves det to doser med 8 ukers intervall som basisvaksinering. Vaksinen mot meningokokk serogruppe B er en nyere vaksine og data om effekt hos stamcelletransplanterte foreligger foreløpig ikke.
Rekvirering av vaksiner til stamcelletransplanterte barn:
| Vaksiner i barnevaksinasjonsprogrammet | Barn opptil 20 år |
| Vaksinene som inngår i barnevaksinasjonsprogrammet, kan bestilles av helsestasjon eller fastlege på vanlig måte (se www.fhi.no). |
| Vaksiner utenom barnevaksinasjonsprogrammet |
|
| Rekvireres fra Folkehelseinstituttet på blå resept § 4 nr. 3 påført indikasjon ‘S’ for stamcelletransplantert pasient |
| Redusert pris til risikogrupper. Hos fastlege før influensasesongen |
| Til pasienter med nedsatt eller manglende miltfunksjon. Rekvireres på blå resept § 4 nr. 3 påført indikasjon ‘M’ for miltmangel (funksjonell eller anatomisk) |
https://www.fhi.no/nettpub/vaksinasjonsveilederen-for-helsepersonell
Oppfølging og etter-kontroll. Senskader
Generelt
Sist faglig oppdatert: 26.05.2020
Hvert år forekommer det i Norge mellom 180–190 nye krefttilfeller hos barn og ungdom yngre enn 18 år. Store forandringer i behandlingsopplegg med bl.a.innføring av stråleterapi og kjemoterapi har ført til en økende andel i voksenbefolkningen som er overlevere etter kreft i barne- og ungdomsalder. Det siste tiåret har utvikling av behandling siktet mot å begrense seneffekter i tillegg til å øke overlevelsen. Frem til nå har få barnekreft-overlevere blitt fulgt gjennom hele livsløpet, slik at vår kunnskap om seneffekter som kan forekomme gjennom et livsløp er begrenset. Stadige endringer av behandlingsopplegg gjør det vanskelig å overføre dagens kunnskap om seneffekter hos gårsdagens barnekreft-overlevere til fremtidige seneffekter hos dagens overlevere. Det foreligger imidlertid stadig mer forskning som viser at så mange som 2 av 3 barnekreftoverlevere lever med en eller flere helsetilstander som kan tilskrives gjennomgått kreftbehandling, i tillegg til ulike sosioøkonomiske utfordringer (Oeffinger et al., 2006). Dette kalles ofte «sen-effekter». Noen sen-effekter utvikler seg allerede under behandlingen («long-term effects»), andre kommer først (flere år) senere («late-effects»).
Barn har større tilpasningsevne og ser dermed ut til å tåle påkjenningene under selve behandlingsfasen bedre enn voksne. Samtidig rammes barn av sykdom og behandling i en alder da de er fysisk og mentalt umodne men i rask vekst og utvikling og derfor er de sårbare for skader på lang sikt. De har også potensielt sett mange flere år igjen å leve sammenlignet med de fleste voksne pasienter.
Når visse organfunksjoner er svekket kan latenstiden før manifestering av symptomer være lang, dette fordi kroppen stiller større krav etter hvert som den vokser til.
Hvilke seneffekter som forekommer, er avhengig av en rekke forhold og samspillet mellom dem ((Kreftoverlevere: ny kunnskap og nye muligheter i et langtidsperspektiv, 2009), tabell 1).
| Forhold ved sykdommen og behandlingen | Type barnekreft. Sykdommens lokalisasjon og utbredelse. Sykdommens vekstmønster. Behandlingsform og behandlingsintensitet. |
| Forhold ved barnet/personen | Alder – jo yngre barnet er, jo mer skadelig er strålebehandling. Resiliens – motstandskraft – noen barn ser ut til å tåle alt. Genetisk utrustning – trolig er visse gener av betydning for utvikling av visse seneffekter. Helseatferd som voksen – røyking, usunn livsstil. |
| Forhold i barnets miljø – av spesiell betydning for psykososiale seneffekter | Familiens måte å tilpasse seg sykdommen. Familiens tilgang på støtte (også profesjonell) inkludert nettverk under behandlingen. Barnets tilgang på støtte (venner, andre voksne og profesjonell). |
(Tabell innhentet fra «Kreftoverlevere: ny kunnskap og nye muligheter i et langtidsperspektiv» (Kreftoverlevere: ny kunnskap og nye muligheter i et langtidsperspektiv, 2009) med tillatelse fra forlaget)
En av utfordringene ved å oppnå kunnskap om disse seneffektene er bl.a. at sykdommen rammer et relativt lite antall barn i ulike aldre og omfatter forskjellige kreftsykdommer i ulike organer, med til dels ulik behandling. En annen viktig utfordring er å bringe kunnskapen ut til allmennhelsetjenesten samt mangel på systematisk oppfølging av overlevere med risiko for seneffekter. Ikke minst er det viktig å bringe kunnskapen om seneffekter ut til pasientene på en måte som gir dem en mulighet til selv å håndtere risikofaktorer for fremtidig helse.
Fysiske seneffekter
Sist faglig oppdatert: 26.05.2020
Tabell 2 viser gradering av risiko for somatiske seneffekter knyttet til forskjellige behandlingsformer (Kreftoverlevere: ny kunnskap og nye muligheter i et langtidsperspektiv, 2009; Wallace et al., 2001).
| Nivå 1: Liten risiko | Kun kirurgisk behandling. Kjemoterapi inkluderte ikke: alkylerende stoffer, anthracycliner, bleomycin eller epipodophyllotoxin. |
| Nivå 2: Moderat risiko | Lave eller moderate doser alkylerende stoffer, anthracycliner, bleomycin eller epipodophyllotoxin. Lave doser av strålebehandling på hjernen. |
| Nivå 3:Høy risiko | All annen strålebehandling Høye doser alkylerende stoffer, anthracycliner, bleomycin eller epipodophyllotoxin. Stamcelle-transplantasjon. |
(Tabell innhentet fra Wallace et al. BMJ 2001 (Wallace et al., 2001) og «Kreftoverlevere: ny kunnskap og nye muligheter i et langtidsperspektiv» (Kreftoverlevere: ny kunnskap og nye muligheter i et langtidsperspektiv, 2009) med tillatelse fra forlag)
Tabell 3 presenterer en summarisk opplisting av mulige seneffekter på forskjellige organsystemer (Dickerman, 2007; Late effects of childhood cancer, 2004), ut i fra organskader, tidligere behandling og andre risikofaktorer, og hvilke tiltak er ønskelige for forebygging og behandling av disse seneffektene.
| Organ | Organskader | Seneffekter | Risikofaktorer i form av behandling | Andre risikofaktorer | Tiltak |
| Sentralnervesystemet | Reduksjon av hvit substans med forkalkninger og tynnere nettverk av kar og nerveceller |
|
|
|
|
| Hormonsystemet | Kreftsykdommen og/eller behandlingen kan påvirke det endokrine organet direkte eller via de overordnete styrende organer |
|
|
|
|
| Pubertet og fertilitet | Jenter:
|
|
|
|
|
|
| Gutter:
|
|
|
|
|
| Hjerte- og karsystemet |
|
|
|
|
|
| Urinveier |
|
|
|
|
|
| Mage-tarm-systemet |
|
|
|
|
|
| Muskel-skjelett-systemet |
|
|
|
|
|
| Lunger |
|
|
|
|
|
| Hørsel |
|
|
|
|
|
| Syn |
|
|
|
|
|
| Tenner og spyttkjertler |
|
|
|
|
|
| Sekundær kreft |
|
|
|
|
|
| Benmargstransplantasjon |
Hud Mage/tarm Lunger Lever Hjernen |
|
|
|
|
Psykososiale seneffekter
Sist faglig oppdatert: 26.05.2020
Generelle forhold
De fleste som overlever kreft i barne- og ungdomsårene ser ut til å klare seg bra senere i livet, uten større grad av psykososiale vansker enn det som finnes i den generelle befolkningen. Overlevere med størst risiko for psykososiale vansker er barn og ungdom behandlet for hjernesvulst, der behandlingen kan føre til forstyrrelser i kognitiv funksjon eller andre nevrologiske seneffekter som epilepsi. Andre risikofaktorer for psykososiale vansker synes å være barnets alder ved diagnose, fysiske seneffekter, antall tilbakefall/behandlingsmengde og barnets og familiens psykologiske ressurser.
Behandlingsbivirkninger som smerter og kvalme samt atskillelse fra familie og venner kan hver for seg representere belastninger som kan føre til psykiske reaksjoner.
Mange som har fått behandling for kreft i ung alder har derimot begrenset hukommelse om forløpet og hva de var igjennom. Enkelte kan faktisk være uvitende om at de har hatt kreft. Ungdom behandlet for kreft synes å være mer sårbare for psykososiale vansker, som igjen kan ha konsekvenser for psykisk og sosial fungering senere i livet, i form av utdanning, yrkesvalg og etablering av egen familie.
Overlevernes familie
Kreftsykdom hos barn/ungdom fører til stor belastning for hele familien. De må leve med sin egen frykt for forløpet og ikke minst utfallet av sykdommen, mestre både opp- og nedturer under behandlingsforløpet og gi barnet omsorg, nærhet og emosjonell støtte. Samtidig er det viktig å opprettholde familiens tidligere funksjon som en enhet. Foreldrenes arbeidsmuligheter og inntekt blir ofte påvirket, de må finne nye måter å fordele oppgaver på seg imellom og deres parforhold kan bli tilsidesatt i kortere eller lengre perioder. Søsken til et kreftsykt barn kan reagere med sjalusi når de får mindre oppmerksomhet eller praktisk oppfølging av foreldrene.
Noen blir rammet av uberettiget skyldfølelse, andre kan reagere med å ta over noe av foreldrenes rolle i hjemmet.
God informasjon og ivaretakelse av foreldrene er derfor viktig og er integrert i behandlingsopplegget på norske barneavdelinger. For fastlegen kan det være viktig å vite at deres pasient har hatt et barn eller et søsken som har gjennomgått kreftsykdom i barnealder. Dette gjelder spesielt de som ikke hadde regelmessig kontakt med avdelingen der barnet/ungdommen ble behandlet (for eksempel søsken). Barneavdelingen tilbyr familien generelle støttetiltak, men ved behov for mer spesielle tiltak som behandling av psykiske plager må dette organiseres av fastlegen.
Sosiale seneffekter
For overlevere av kreft i barne- eller ungdomsårene ser de fleste ut til å klare seg som sine jevnaldrende i forhold til utdanning, arbeidsliv og etablering av egen familie. Like vel synes noen grupper å ha større risiko for dårligere funksjon i skole. Barn behandlet for hjernesvulster, skiller seg ut som den mest utsatte gruppen grunnet de tidligere nevnte kognitive og nevrologiske seneffektene. Også barn med akutt leukemi som har fått cellegift intratekalt eller blitt bestrålt, klarer seg ofte dårligere på skolen. Disse barna trenger ekstra tiltak eller spesialundervisning i større grad sammenlignet med sine søsken. Dette administreres av skolen og kommunen men må basere seg på legenes kunnskap om sammenhengen mellom skolevansker, sykdom og behandling, og om plagene vil være forbigående eller økende over tid.
Rapporter angående sosial tilpasning blant overlevere av kreft i barnealder og ungdom har vist noe motstridende funn, men materialene har vært små. Nordiske data viser at færre kreftoverlevere er i arbeid og flere mottar trygd enn kontroller fra den generelle befolkningen. Det er usikkert om dette funnet gjenspeiler psykiske plager eller somatiske seneffekter, eller om det er uttrykk for at det er lavere terskel for å søke om trygdeytelser etter gjennomgått kreftsykdom. Økende forekomst av somatiske seneffekter med økende alder kan også føre til at enkelte overlevere vil ha mer sykefravær eller forbli kortere i arbeidslivet. Riktige tiltak vil oftest være sammensatte med utgangspunkt i individuelle behov.
En norsk studie av alle i alderen 17–44 år i perioden 1974–2001, inkluderte 12,100 personer med gjennomgått kreft (både i barne- og ung voksen alder), fant at det ikke var signifikant forskjell i andelen gifte i kreftgruppen sammenlignet med den kreftfrie kontrollgruppen, bortsett fra en lavere ekteskapsrate for kvinnelige overlevere etter hjerne- eller brystkreft, og ingen store avvik for mennene (Syse, 2008). En ny norsk studie av mannlige overlevere etter barne-og ungdomskreft født i perioden 1965–1985, viste en noe reudsert ekteskapsrate, særlig for overlevere etter CNS tumores. Den viste også at de mannlige barnekreftoverleverne i mindre grad enn kontrollene fikk egne barn (nær 30 % redusert sannsynlighet for å få egne barn), samt økt bruk av in vitro fertilisering (tre ganger økt risiko), men ingen økning i negative utfall for deres førstefødte (dødfødsel, prematuritet, dysmaturitet eller medfødte misdannelser) (Gunnes et al., 2016).
Psykiske seneffekter
Ut i fra dagens kunnskap har man ikke kunnet påvise økt forekomst av psykiske lidelser hos overlevere av kreft i barnealder/ungdom, med unntak av psykoser etter hjernesvulster. Noen studier har vist økt forekomst av symptomer på PTSD («post traumatic stress disorder»), engstelighet og nedstemthet. Symptomer som redsel for tilbakefall, angst for egen helse og somatisering er lite studert, men for noen kan en vedvarende bekymring for helsen bli et problem. Større undersøkelser av helseatferd (f.eks røyking eller mosjon) viser at de fleste overlevere av kreft i barne- og ungdomsårene har en sunn livsstil og andel røykere blant dem er lavere enn blant deres jevnaldrende.
Det er viktig at leger og annet helsepersonell som behandler kreftoverlevere er klare over muligheten for psykiske plager og at de gir overleveren mulighet til å ta opp disse plagene. Ved å normalisere plagene kan det ofte være nok å kartlegge og gi enkle råd. Det kan medføre at bekymringen for plagen reduseres samtidig som overleverens egen mestring av situasjonen kan støttes. Hvis det foreligger psykiatriske lidelser skal disse behandles av det psykiske hjelpeapparatet. Flere nyere forskningsrapporter finner økt forekomst av kronisk utmattelse (fatigue) hos overlevere etter barnekreft (Hamre et al., 2012). Tilstanden har likheter med «Kronisk fatigue syndrom/ME» men er ofte mindre uttalt.
Oppfølging
Sist faglig oppdatert: 26.05.2020
Kontroller for og forebygging av seneffekter etter kreft i barnealder/ungdom er viktig. Det reiser store krav til helsevesenet av organisatorisk og faglig karakter.
Oppfølging av barn
Barn som har fått behandling for en kreftsykdom, følges vanligvis opp ved barneavdelingene frem til 18 års alder. Tidlige seneffekter blir som oftest fanget opp der og behandling iverksatt. Størst risiko for tidlige seneffekter har barn og ungdom som er behandlet for hjernesvulster. Disse seneffektene kan være fysiske og forårsaket av mangel på vekst- eller kjønnshormoner. Nevrologiske eller nevropsykologiske seneffekter i form av problemer med kognitive funksjoner som hukommelse, konsentrasjon og orientering, kan ofte bli merkbare flere år etter at behandlingen er avsluttet. Disse overlevere trenger tett oppfølging av mange instanser etter avsluttet behandling.
Ved 18 års alder blir overlevere med behov for videre oppfølging som regel overført til og fulgt opp av respektive spesialistavdelinger. Andre får en informasjonssamtale ved avsluttende kontroll og henvises videre til fastlegen.
Oppfølging av voksne
Eksisterende data tyder på at majoriteten av seneffektene vil vise seg først opp til 10–40 år etter avsluttet behandling og dermed være ukjente for behandlende lege og for overleveren når kontrollene på barneavdelingen avsluttes. Videre oppfølging av barnekreft-overlevere med økt risiko for å utvikle alvorlige seneffekter er et uløst problem i vårt helsevesen men har fått økt fokus de siste årene.
Helsedirektoratet nedsatte i mars 2010 en arbeidsgruppe som skulle vurdere grunnlaget for systematisk oppfølging av kreftoverlevere. I gruppens rapport presenteres en modell hvor kreftoverlevere stratifiseres til tre risikonivåer (Oppfølging av kreftoverlevere med særlig fokus på seneffekter: innstilling fra en arbeidsgruppe nedsatt av Helsedirektoratet, 2010). Nivå 1 representerer gruppen med liten risiko for seneffekter og de utgjør ca 75 % av alle kreftoverlevere. Disse kan generelt følges opp av primær-helsetjenesten. Nivå 2 inkluderer gruppen med moderat risiko for seneffekter. Denne gruppen utgjør ca 20 % av overleverne og de kan følges primært opp i allmennhelsetjenesten men henvises til spesialisthelsetjenesten ved behov. Siste 5 % av kreftoverleverne tilhører nivå 3 med høy risiko for seneffekter. Disse trenger vedvarende oppfølging av spesialisthelsetjenesten i samarbeid med primærhelsetjenesten, Det finnes per i dag ingen spesifikke instanser (foruten en voksenpoliklinikk på St Olavs Hospital) hvor disse kan henvises. Ved å opprette en seneffektpoliklinikk vil man kunne etablere et nettverk av spesialister med interesse for og kunnskap om seneffekter, slike sen-effekt poliklinikker er godt etablert i andre europeiske land som f.eks Nederland og Portugal.
Fastlegene må bli informert om risikofaktorer og helseproblemer som overlevere av barnekreft er utsatt for, både generell kunnskap men ikke minst individuelle opplysninger for de overleverene de behandler. Et skjema med informasjon om tidligere behandling og mulige risikofaktorer (et såkalt «survivor passport») er under arbeid (PanCare, et europeisk samarbeid). Det skal følge overleveren (som skal «eie» dokumentet) når oppfølging på barneavdeling avsluttes.
Overleverene må også ta sin del av ansvaret. De har rett på informasjon om fremtidig risiko for helseproblemer for å kunne styre sin atferd slik at adderende risikofaktorer minimaliseres. Tidspunktet for når overlevere av kreft i barndom og ungdom er mottagelige for den type informasjon er ikke sikkert, men studier har vist at flertallet av overlevere som nå er i voksen alder, har minimal kunnskap om senvirkningene av sin behandling.
Nevropsykologisk oppfølging etter behandling for hjernesvulst
Sist faglig oppdatert: 26.05.2020
Kognitiv funksjon
Operasjon og sykdomsforløp er belastende for barn/ungdom og deres familie og nettverk. Når barnet/ungdommen må gjennom behandling etter operasjonen i form av stråling eller cytostatika, øker dessverre sjansene for kognitive senskader (Levisohn, Cronin-Golomb, & Schmahmann, 2000; Mulhern, 2003; Mulhern & Butler, 2004). Det kan bli flere sykehusinnleggelser, og fraværet fra hjem og barnehage/skole kan bli langvarig. I tillegg medfører selve behandlingen ofte at barnet blir hengende etter i skolefagene og i lek og samspill med jevnaldrende.
Avgjørende for art og grad av følgevirkninger er:
- barnets alder
- svulsttype og beliggenhet
- type etterbehandling
Senskader etter strålebehandling
Barn som gjennomgår strålebehandling, kan få lærevansker av ulik grad (Mulhern & Butler, 2004). Dette oppstår fordi den behandlingen som skal virke på svulstvevet og hindre tilbakefall, også påvirker de friske delene av hjernen. Det kan dreie seg om konsentrasjons-, innlærings-, og hukommelsesproblemer, eller vansker med å forholde seg til flere ting på en gang. På sikt kan dette gi problemer med å ta til seg ny lærdom, slik at barnet sakker akterut på skolen. Noen ganger oppstår økt trettbarhet og humørsvingninger som følge av behandlingen. Samlet kan dette gi både faglige og sosiale problemer på skolen, men også i hverdagsliv og fritid.
Senskader etter cytostatikabehandling
Noen av de forannevnte vanskene kan også forekomme etter kun cellegiftbehandling, men da som regel i mildere grad.
Senskader etter operasjon
Man antok tidligere at operasjonen ikke hadde langtidseffekt på kognitiv og psykososial funksjon. Flere nyere studier har imidlertid vist at også i denne gruppen forekommer senskader, selv om art og grad er mer variert enn i gruppen som trenger etterbehandling (Rønning, Sundet, Due-Tønnessen, Lundar, & Helseth, 2005; Aarsen, Van Dongen, Paquier, Van, & Catsman-Berrevoets, 2004).
Oppfølging
For å kunne avdekke problemer som kan oppstå og gi hjelp underveis, bør alle barn og familier få tilbud om nevropsykologisk og pedagogisk kartlegging på bestemte tidspunkt. Dette gir grunnlag for anbefalinger om tilrettelegging i skole og barnehage. Både barn, foreldre og søsken vil noen ganger ha behov for støttesamtaler hos kyndig personell om de følelsesmessige og sosiale følgevirkningene som kan oppstå.
Det fins mange muligheter for tilrettelegging og trening etter operasjon og behandling for hjernesvulst. Det er viktig å utarbeide tiltakene i samarbeid med barnas skole og nærmiljø. Nyere studier har vist positive virkninger av enkelte kognitive treningsprogrammer (Hagberg-van't Hooft, 2005), men effekt av direkte trening av kognitive ferdigheter er enda lite dokumentert.
Det er satt i gang et felles oppfølgingsprogram for ivaretakelse av barna og familiene ved alle regionssykehusene. Det har ved Rikshospitalet vist seg formålstjenlig å undersøke barna én gang tidlig i sykdomsforløpet, og så cirka ett, to og fem år etter avsluttet behandling.
Palliativ og terminal behandling
Sist faglig oppdatert: 26.05.2020
Det at barn dør av uhelbredelig kreftsykdom eller annen sykdom, er heldigvis sjelden. Dette kapittelet har til hensikt å beskrive de behandlingsoverveielser som bør tas ved behov for lindrende behandling i terminalfasen hos barn. Prosessene rundt beslutningen om å gå fra kurativt rettet behandling til behandling kun med mål lindring tas ikke opp her.
Lindrende behandling i terminalfasen, både hos barn og voksne, må individualiseres. Retningslinjene her er primært utarbeidet med tanke på barn med kreftsykdom, men bør kunne overføres også til andre sykdomsgrupper.
Som hovedregel bør det være barnets faste leger som er ansvarlige for den siste delen av behandlingen i de tilfellene hvor barn dør etter et terminalt sykeleie. Det er de som har kjent familien gjennom lang tid, som har best forutsetninger for å trekke opp linjene for behandlingen også i den terminale fasen.
Gjennomføringen av terminal behandling er basert på god dialog mellom lege, pasient og dennes foresatte. Da kan en i størst mulig grad legge opp behandlingen på pasientens premisser. Ved behandling av barn forholder man seg til en hel familie, og det er alltid en meget tung og langvarig prosess for foreldrene å innse og akseptere at barnets liv ikke står til å redde. Dette kan influere på de siste faser i behandlingen av barn, ved at det oftest tar lengre tid før man kan konsentrere seg om symptomlindring alene.
Terminal sedering til barn er erfaringsmessig mindre aktuelt. Dette fordi det oftest settes som et overordnet mål å opprettholde en bevisst kontakt mellom barn og foreldre så lenge som mulig.
Det har vært drøftet om terminal sedering ut fra et etisk perspektiv kan inngå som en modalitet i lindrende behandling. I den grad man har oppnådd konsensus, kan en si at her, som ellers i medisinen, er det styrende prinsipp at det som er det beste for pasienten, er det som vektlegges mest i valg av behandling. I gitte tilfeller kan derfor terminal sedering være aktuelt også til barn.
Pasienter i terminalfase av kreftsykdom skal vanligvis ikke utsettes for cardiopulmonal resuscitering. Dette må nedfelles tydelig i journalen. Fra referanse 166:
«Beslutninger om å begrense livsforlengende behandling skal dokumenteres. Det bør angis når ny evaluering skal finne sted. Dokumentasjonen må omfatte hvilken behandling som skal og ikke skal gis, det medisinske grunnlaget for beslutningen, hvilken informasjon som er gitt til pasient og pårørende, samt pasientens ønske.»
Hvis spørsmålet om terminal sedering kommer opp, bør beslutninger om dette tas i et bredt kollegium. Spesialister på området bør med lav terskel konsulteres.
Alle beslutninger om behandling i terminalfasen må skje i full åpenhet med pasientens foreldre og pasienten selv hvis han/hun er moden nok til å forstå.
Elementer ved den praktiske gjennomføringen
Hovedmålsetningen ved den terminale behandlingen er symptomlindring.
Hvis familien ønsker det, bør barnet få være hjemme. Terminalbehandlingen ivaretas da i et samarbeid mellom behandlende barneavdeling (leger og sykepleiere) og hjemmesykepleien, eventuelt andre instanser (f.eks. Franziskushjelpen). Det er som regel ønskelig at sykepleiere fra behandlende barneavdeling kan reise på hjemmebesøk etter nærmere avtale, ta eventuelle blodprøver, administrere intravenøse medisiner og eventuelt væske.
Ta så lite prøver som mulig!
Cytostatika og steroider brukes i noen grad palliativt etter individuell vurdering.
Optimal smertelindrende behandling skal gis etter behov. Se eget punkt nedenfor
Husk behandling av obstipasjon ved langvarig bruk av opioider.
Transfusjoner gis etter individuell vurdering, og erytrocytter ses sjelden som indisert. Hos barn som er mest hjemme, ønsker en ofte å holde trombocyttverdier slik at risikoen for akutte store blødninger er liten (dvs: trombocytter > ca. 10).
Benzodiazepiner (diazepam) kan gis for å lindre angst og uro og eventuelt for å bedre effekten av analgetika.
Hvis barn med terminal kreftsykdom må legges inn på sykehus, skal de som hovedregel ligge der de er kjent fra tidligere. Barn på sykehus trenger ofte kontinuerlig morfindrypp som smertestillende behandling. Øk dosen gradvis til smertelindring oppnås. Husk kombinasjonsbehandling med paracetamol og eventuelt NSAIDs. Blokkader med lokalanestetikum kan gjerne lettere administreres og vedlikeholdes på sykehuset, og bør vurderes hvis antatt å være til hjelp.
Benzodiazepiner (midazolam mest aktuelt ved behov for kontinuerlig infusjon) kan brukes som supplement til morfininfusjon hvis nødvendig. Balanser graden av symptomlindring mot ønsket våkenhet. Behandling gis evt i samråd med spesialister på lindrende behandling/smertebehandling
Respirasjonsbesvær kan lindres ved oksygentilførsel, men oksygentensjonen skal ikke måles rutinemessig hvis barnet ikke er besværet.
Behandling av infeksjoner, væsketilførsel og ernæring vurderes ut fra forventet levetid og om forventet lindring.
Om smertebehandlingen
De fleste (men ikke alle) barn som dør av kreft, har smerter og behov for smertelindrende behandling. Ved mange kreftsykdommer bruker man en mild cytostatikabehandling som palliativ behandling når kurativ behandling ikke lenger er mulig. Palliativ strålebehandling kan også være et godt alternativ ved vanskelig traktable smerter ved uhelbredelig kreftsykdom. Ofte(st) vil det ha vært behov for smertelindrende behandling allerede i lang tid før barnet går over i det som kan kalles terminal fase. Det vil da som regel være instituert behandling med opioider, enten i form av et kombinasjonspreparat med paracetamol, som et langsomtvirkende morfinpreparat, depotplaster (fentanyl, buprenorfin) eller eventuelt metadon mikstur. På grunn av toksiske metabolitter bør petidin og dekstropropoksyfen unngås ved behandling av kreftrelatert smerte (Retningslinjer for smertelindring, 2009). Skifte av opioider, eventuelt rotasjon av ulike opioider, kan forsøkes hvis en har vansker med store bivirkninger eller ujevn smertelindring. Lokale blokkader med lokalanestetikum kan være aktuelt, og bør vurderes hvis tilstanden ligger til rette for det. I terminal fase må dosene oftest økes for å oppnå / vedlikeholde smertefrihet, ofte langt høyere enn vanlig anbefalte doser. Å gi opioidene intravenøst eller subcutant, gjerne i form av kontinuerlig infusjon, vil kunne være beste administrasjonsform. Oftest gis opioidene i kombinasjon med paracetamol eller ikke-steroide antiinflammatoriske medikamenter. Hvis dosene av opioidene må økes raskt for å oppnå tilfredsstillende smertelindring, kan somnolens og respirasjonsdepresjon være bivirkninger. Somnolens i denne situasjonen kan være hensiktsmessig. Respirasjonsdepresjon vil man prøve å unngå ved langsom doseøkning og kombinasjonsbehandling med andre typer analgetika. Balansegang mellom bivirkninger og de ønskede smertestillende effekter kan gjøre doseringen vanskelig, siden begge er doseavhengige. En del kreftformer kan utløse nevropatisk smerte hvor tilnærmingen atskiller seg fra nociceptiv smertebehandling.
Ved terminal lindring hvor smerter utgjør store plager for pasienten, må symptomlindring settes som overordnet målsetning. «Når livsforlengende behandling avsluttes, skal lindrende behandling videreføres, eller trappes opp. Pasienten skal ha adekvat smertebehandling, selv om det ikke kan utelukkes at dette framskynder døden.» (2013).
Annen form for lindrende behandling
Bevisstheten blir ofte redusert i terminalfasen. Denne fasen er ofte ledsaget av angst, pustebesvær, diverse ukarakteristiske symptomer, eventuelt kramper. Det viktigste angstdempende tiltaket er at barnet har sine nærmeste hos seg, og at disse igjen har den nærhet og støtte som de behøver for å klare å takle situasjonen. Angstdempende medikamenter kan likevel bli nødvendig og vil også kunne bedre den subjektive effekten av analgetika. Benzodiazepiner er best egnet til dette bruk. Dette er også den medikamentgruppen man bruker for å stoppe kramper.
Pustebesvær kan være et symptom i terminalfasen og er i tilfelle meget angstskapende. Oksygen skal kun brukes hvis det lindrer barnets subjektive besvær. I denne fasen bør man vanligvis ikke måle oksygenmetning, og det å gi oksygen for å heve en målt lav SaO2 uten ledsagende plagsomme symptomer, er kontraindisert. Også i denne situasjonen kan benzodiazepiner lindre symptomer. Opioider, eventuelt i kombinasjon med benzodiazepiner, er en god og skånsom lindrende behandling av uro forårsaket av lufthunger, selv om det kan medføre hypoventilasjon.
Prosess og metode for utarbeiding av retningslinjene
Hva er nasjonale retningslinjer?
Sist faglig oppdatert: 26.05.2020
I henhold til Nasjonal helseplan (2007–2010) (Nasjonal helseplan (2007-2010), 2006) har Helsedirektoratet en normerende rolle for helsetjenesten på tvers av helseregioner og tjenestenivå. Helsedirektoratet har en koordinerende rolle for å utvikle overordnede referanserammer for kreftomsorgen, sammen med de regionale helseforetakene, kommunene og andre relevante myndighetsorganer og tjenester.
Lov om kommunale helse- og omsorgstjenester § 12-5 fastslår at Helsedirektoratet er eneste aktør med mandat til å utvikle, formidle og vedlikeholde nasjonale faglige retningslinjer og veiledere som understøtter de mål som er satt for helse- og omsorgstjenesten.
De nasjonale faglige retningslinjene inneholder systematisk utviklede faglige anbefalinger som etablerer en nasjonal standard for utredning, behandling og oppfølging av pasientgrupper, diagnosegrupper og brukergrupper. Nasjonale faglige retningslinjer er et virkemiddel for å bidra til at helse- og omsorgstjenestene:
- har god kvalitet
- gjør riktige prioriteringer
- ikke har uønsket variasjon i tjenestetilbudet
- løser samhandlingsutfordringer
- tilbyr helhetlige pasientforløp
Nasjonale faglige retningslinjer gir uttrykk for hva som anses som god praksis på utgivelsestidspunktet. Sentrale fagmiljøer og tjenestemottakere er aktivt involvert i utarbeidelsen.
Helsepersonell og alle deler av helse- og omsorgstjenesten er forpliktet til å yte forsvarlig helsehjelp. Retningslinjer, anerkjent fagkunnskap og allmenngyldige samfunnsetiske normer inngår som aksepterte grunnlag for vurdering av hva som er faglig forsvarlig. Retningslinjer er ment som et hjelpemiddel ved avveiningene tjenesteyterne må gjøre for å oppnå forsvarlig og god kvalitet i tjenesten. De er ikke rettslig bindende, men faglig normerende for valg man anser fremmer kvalitet, god praksis og likhet i tjenesten på utgivelsestidspunktet. Helsepersonell må likevel vise faglig skjønn i vurderingen av hver enkelt pasient for å ta hensyn til individuelle behov. Dersom helsepersonell eller institusjoner velger å fravike anbefalinger i en retningslinje, skal dette dokumenteres og begrunnes.
Helsetjenestens eiere og ledelse har ansvar for tilrettelegging av virksomheten slik at anbefalinger gitt i nasjonale faglige retningslinjer kan følges.
Kunnskapsbasert prosess
Sist faglig oppdatert: 26.05.2020
Helsedirektoratet legger til grunn at alle nasjonale retningslinjer skal være utarbeidet etter en metode med vekt på forskningsbasert kunnskap, tydelig og tilgjengelig dokumentasjon, brukermedvirkning, tverrfaglighet, fokus på praksis, implementering og oppdatering.
Det er en omfattende prosess å lage gode retningslinjer som tilfredsstiller krav til prosess, metode og transparens som er det nivå Helsedirektoratet og andre internasjonale organisasjoner har lagt til grunn for utforming av anbefalinger.
Helsedirektoratet ønsker i arbeidet med nasjonale retningslinjer for kreftbehandling å bygge på det arbeid onkologiske faggrupper i en årrekke har gjort med å lage behandlingsveiledere/ handlingsprogram.
I dette Nasjonale handlingsprogrammet med retningslinjer for diagnostikk, behandling og oppfølging av barnekreft har faggruppen i hovedsak vektlagt internasjonalt aksepterte behandlings- og forskningsprotokoller som grunnlag for behandlingsanbefalinger og litteraturbakgrunn. Dette aksepteres fordi nevnte protokoller utarbeides på en måte som tilsvarer den kunnskapsbaserte prosessen som man ønsker skal ligge til grunn for et handlingsprogram.
Gradering av kunnskapsgrunnlaget
Sist faglig oppdatert: 26.05.2020
Ved utarbeiding av nasjonale retningslinjer stilles det krav om at all relevant kunnskap på området hentes frem, beskrives og vurderes på en systematisk og åpen måte. I dette handlingsprogrammet har man ikke funnet dette hensiktsmessig da det har vært mer naturlig til å referere til de ulike protokoller. Der finnes oppsummering og henvisninger til all sentral litteratur.
I disse retningslinjene har man derfor kun i noen grad benyttet seg av følgende graderingsmodell for å vise hvilket vitenskapelig grunnlag kunnskapen er basert på.
| Studietype | Gradering av evidensnivå |
|---|---|
| Kunnskap som bygger på systematiske oversikter og meta-analyser av randomiserte kontrollerte studier. Kunnskap som bygger på minst én randomisert kontrollert studie | A |
| Kunnskap som bygger på minst én godt utformet kontrollert studie uten randomisering Kunnskap som bygger på minst én annen godt utformet kvasi-eksperimentell studie uten randomisering | B |
| Kunnskap som bygger på godt utformede ikke eksperimentelle beskrivende studier, som sammenlignende studier, korrelasjonsstudier og case studier | C |
| Kunnskap som bygger på rapporter eller oppfatninger fra eksperter, komiteer og/eller klinisk ekspertise hos respekterte autoriteter | D |
Bakgrunn og arbeidsprosess
Sist faglig oppdatert: 26.05.2020
Utvikling av nasjonale handlingsprogrammene for kreftbehandling er et viktig tiltak under Nasjonal strategi for kreftområdet (2006–2009). Nasjonale handlingsprogrammer for kreftbehandling skal bidra til at det offentlige tilbudet i kreftomsorgen blir av god kvalitet og likeverdig over hele landet.
Nasjonale faggrupper innen onkologi har i en årrekke arbeidet med og utviklet behandlingsveiledere og handlingsprogram for ulike krefttyper. Helsedirektoratet ønsket i arbeidet med nasjonale handlingsprogrammer som ledd i Nasjonal strategi for kreftområdet å ta utgangspunkt i og bygge på dette arbeidet. Helsedirektoratet rettet derfor i 2009 en henvendelse til fagmiljøet innen barneonkologi og ba om forslag til representanter til en arbeidsgruppe som skulle settes sammen av fagekspertise, Nasjonalt kunnskapssenter for helsetjenesten og Helsedirektoratet. Det ble bedt om at alle nødvendige faggrupper skulle være representert, og at gruppen skulle bestå av fagfolk fra alle helseregioner.
RHF’ene har medvirket i arbeidet gjennom representasjon i prosjektets styrings- og referansegruppe, samt ved mulighet til å gi tilbakemelding på arbeidsgruppenes sammensetning. RHF’ene ble også bedt om at fagfolk innenfor rammen av sin arbeidstid ble fristilt til retningslinjearbeidet, jf. Oppdragsdokumentet fra Helse- og omsorgsdepartementet.
Første versjon av handlingsprogrammet utgitt 27.06.2014 (IS-1868)
Det første Nasjonale handlingsprogrsammet for kreft hos barn ble skrevet av en arbeidsgruppe bestående av følgende personer:
- Marit Hellebostad, Barneklinikken, Oslo universitetssykehus Ullevål, nå barneavdelingen, Vestre Viken HF (leder)
- Kristin Bjørnland, barnekirurgisk seksjon, avdeling for lever, gastro og barnekirurgi, Klinikk for spesialisert medisin og kirurgi, OUS – Rikshospitalet
- Johan Cappelen, Nevrokirurgisk avdeling, St. Olavs Hospital HF, Trondheim
- Øystein Drivenes, Barnekirurgisk avdeling, St. Olavs Hospital HF, Trondheim
- Bernt J. Due-Tønnessen, Nevrokirurgisk avdeling, Seksjon for barnenevrokirurgi Oslo universitetssykehus, Rikshospitalet
- Odd Monge, Onkologisk avdeling, Haukeland Sykehus HF, Bergen
- Tore Stokland, tidligere leder av faggruppe for svulster i sentralnervesystemet hos barnBarneavdelingen, Universitetssykehuset Nord Norge, Tromsø
- Finn Wesenberg, tidligere leder av kompetansesenteret for solide svulster hos barn, Oslo universitetssykehus Rikshospitalet
- Eva Widing, tidligere leder av faggruppe for solide svulster utenfor sentralnervesystemet hos barn, Barnemedisinsk avdeling, Oslo universitetssykehus Rikshospitalet
- Bernward Zeller, leder av kompetansesenteret for solide svulster hos barn, Oslo universitetssykehus Rikshospitalet
- Ann Elisabeth Åsberg, Barneklinikken, St. Olavs Hospital HF, Trondheim
Øvrige forfattere
- Kjell Magnus Antonsen, psykologspesialist, Barneklinikken, Oslo universitetssykehus Ullevål
- Klaus Beiske, avdeling for patologi, Oslo universitetssykehus Rikshospitalet
- Torhild Berntsen, Barneavdeling for nevrofag, Oslo universitetssykehus Rikshospitalet
- Petter Brandal, Avdeling for kreftbehandling, Oslo universitetssykehus Radiumhospitalet
- Jochen Büchner, Barnemedisinsk avdeling, Oslo universitetssykehus Rikshospitalet
- Trond Flægstad, Barneavdelingen, Universitetssykehuset Nord Norge, Tromsø
- Alexander Fosså, Avdeling for kreftbehandling, Oslo universitetssykehus Radiumhospitalet
- Anders Glomstein, Barnemedisinsk avdeling, Oslo universitetssykehus Rikshospitalet
- Heidi Glosli, Barnemedisinsk avdeling, Oslo universitetssykehus Rikshospitalet
- Ketil Heimdal, avdeling for medisinsk genetikk, Oslo universitetssykehus Rikshospitalet
- Thore Henrichsen, Barneklinikken, Oslo universitetssykehus Ullevål, nå Barneavdelingen, Vestre Viken HF
- Inga María Rinvoll Jóhannsdóttir, Nasjonalt kompetansesenter for seneffekter, Avdeling for kreftbehandling, Oslo universitetssykehus Montebello og Barnemedisinsk avdeling, Oslo universitetssykehus Rikshospitalet
- Adriani Kanellopoulos, Barneavdelingen, Akershus universitetssykehus
- Per Kristian Knudsen, Barneklinikken, Oslo universitetssykehus Ullevål
- Knut Lote, onkologisk avdeling, Oslo universitetssykehus Radiumhospitalet
- Bendik Lund, Barneklinikken, St. Olavs Hospital HF, Trondheim
- Randi Nygaard, Barneklinikken, St. Olavs Hospital HF, Trondheim
- Sidsel Randklev, sosionom, Barneklinikken, Oslo universitetssykehus Ullevål
- Wenche Reed, seksjonsleder, Stab forskning, innovasjon og utdanning, Oslo universitetssykehus
- Ellen Ruud, Barnemedisinsk avdeling, Oslo universitetssykehus Rikshospitalet
- Einar Stensvold, Barnemedisinsk avdeling, Oslo universitetssykehus Rikshospitalet
- Randi Skarpaas Tranheim, øyeavdelingen, Oslo universitetssykehus Ullevål
- Ingjerd Aagenæs, Barneradiologisk enhet, Avd. for radiologi og nukleærmedisin, Oslo universitetssykehus HF, Ullevål
Arbeidsgruppen leverte i desember 2010 første utkast til Nasjonalt handlingsprogram med retningslinjer for diagnostikk, behandling og oppfølging av barnekreft. Etter tilbakemelding fra Helsedirektoratet på utkastet april 2011 mottok Helsedirektoratet i januar 2012 et revidert utkast til nasjonalt handlingsprogram. Faggruppen, Nasjonalt kunnskapssenter for helsetjenesten og Helsedirektoratet samarbeidet i 2012 til mai 2013 om videre ferdigstillelse av handlingsprogrammet og det ble i mai 2013 sendt på høring til:
- Faggruppen for svulster i sentralnervesystemet hos barn
- Faggruppen for solide svulster utenfor CNS hos barn og barneleukemigruppen
- Kreftforeningen
- Støtteforeningen for kreftsyke barn (nå: Barnekreftforeningen)
- De regionale helseforetakene
- Den norske legeforening
Faggruppen og Helsedirektoratet samarbeidet høsten 2013 til våren 2014 om ferdigstillelse av handlingsprogrammet.
Andre versjon av handlingsprogrammet - utgitt 03.09.2015 (IS-2366)
Forfattere
- Marit Hellebostad, Barneklinikken, Oslo universitetssykehus Ullevål, nå Barneavdelingen, Vestre Viken HF (leder)
- Kristin Bjørnland, barnekirurgisk seksjon, avdeling for lever, gastro og barnekirurgi, Klinikk for spesialisert medisin og kirurgi, Oslo universitetssykehus HF, Rikshospitalet
- Johan Cappelen, Nevrokirurgisk avdeling, St. Olavs Hospital HF
- Øystein Drivenes, Barnekirurgisk avdeling, St. Olavs Hospital HF
- Bernt J. Due-Tønnessen, Nevrokirurgisk avdeling, Seksjon for barnenevrokirurgi Oslo universitetssykehus HF, Rikshospitalet
- Odd Monge, Onkologisk avdeling, Haukeland universitetssykehus HF
- Tore Stokland, leder faggruppe for svulster i sentralnervesystemet hos barn, Barneavdelingen, Universitetssykehuset Nord Norge HF
- Finn Wesenberg, leder kompetansesenteret for solide svulster hos barn, Oslo universitetssykehus HF, Rikshospitalet
- Eva Widing, leder faggruppe for solide svulster utenfor sentralnervesystemet hos barn, Barnemedisinsk avdeling, Oslo universitetssykehus HF, Rikshospitalet
- Bernward Zeller, Barnemedisinsk avdeling, Oslo universitetssykehus HF, Rikshospitalet
- Ann Elisabeth Åsberg, Barneklinikken, St. Olavs Hospital HF
(Øvrige delforfattere omtales i kapittel 12 Prosess og metode)
Nye forfattere i revidert utgave utgitt 03.09.2015
- Monica Cheng Munthe-Kaas, Barnemedisinsk avdeling, Oslo universitetssykehus Rikshospitalet
- Jochen Büchner, Barnemedisinsk avdeling, Oslo universitetssykehus Rikshospitalet
- Margrete Greve-Isdahl, Nasjonalt Folkehelseinstitutt, Oslo
- Maria Winther Gunnes, Barneklinikken, Haukeland Universitetssykehus
- Forskerne Ingvil Sæterdal og Lene Juvet, forsker, Nasjonalt kunnskapssenter for helsetjenesten har bistått arbeidsgruppen med kunnskapssøk og gradering av kunnskapsgrunnlaget.
Tredje versjon av handlingsprogrammet - utgitt 10.01.2017 (IS‑2586)
Revisjonsgruppe 2016
- Maria Winther Gunnes (leder), Barneklinikken, Haukeland Universitetssykehus, Bergen
- Bernt J. Due-Tønnessen, Nevrokirurgisk avdeling, Seksjon for barnenevrokirurgi Oslo universitetssykehus, Rikshospitalet
- Bernward Zeller, leder av kompetansesenteret for solide svulster hos barn, Oslo universitetssykehus Rikshospitalet
- Niklas Stabell, Barneavdelingen, Universitetssykehuset Nord Norge, Tromsø
Revisjonsgruppen sendte juni 2016 revidert Nasjonalt handlingsprogram med retningslinjer for diagnostikk, behandling og oppfølging av barnekreft, til høring til lederne for:
- Faggruppen for svulster i sentralnervesystemet hos barn
- Faggruppen for solide svulster utenfor CNS hos barn
- Norsk barneleukemigruppe
Endelig revidert versjon ble sendt Helsedirektoratet 27. november 2016 og Nasjonalt handlingsprgram ble utgitt av Helsedirektoratet 10.01.2017.
Hovedforfattere av de enkelte kapitler
|
| Opprinnelig(e) forfatter(e) | Revisjon 2016 |
|---|---|---|
| 1 Innledning | Bernward Zeller og Finn Wesenberg | Bernward Zeller |
| 2 Epidemiologi | Bernward Zeller og Finn Wesenberg | Bernward Zeller |
| 3 Forebygging |
|
|
| 4 Forløpstider |
| Helsedirektoratet |
| 4.1 Om pakkeforløp for kreft |
|
|
| 4.2 Forløpstider for kreft hos barn |
|
|
| 5 Tidlig diagnostikk/screening |
|
|
| 6 Diagnostisering |
|
|
| 6.1 Symptomer, utredning, stadieinndeling | Bernward Zeller | Bernward Zeller |
| 6.2 Patologisk-anatomisk diagnostikk av barnekreft | Klaus Beiske | Klaus Beiske |
| 6.3 Integrert diagnostikk |
|
|
| 6.4 Biobank | Wenche Reed | Wenche Reed |
| 7 Genetikk |
|
|
| 7.1 Arv og kreft hos barn | Ketil Heimdal | Kjetil Heimdal |
| 7.2 Molekylærgenetiske forhold | Kjetil Heimdal | Kjetil Heimdal |
| 8 Behandling |
|
|
| 8.1 Generelt om medikamentell behandling | Bernward Zeller | Bernward Zeller |
| 8.2 Kirurgi |
|
|
| 8.2 - Avsnitt "Kirurgisk behandling av solide svulster utenfor sentralnærvesystemet" | Kristin Bjørnland og Øystein Drivenes | Kristin Bjørnland og Øystein Drivenes |
| 8.2 - Avsnitt "Kirurgisk behandling av svulster i sentralnervestystemet" | Bernt J. Due-Tønnessen | Bernt J. Due-Tønnessen |
| 8.3 Strålebehandling | Petter Brandal, Knut Lote og Odd Monge | Petter Brandal |
| 8.4 Utprøvende behandling | Finn Wesenberg | Bernward Zeller/Maria W. Gunnes |
| 8.5 Akutte onkologiske tilstander | Anne Elisabeth Åsberg | Bernward Zeller, Maria W. Gunnes, Bernt Due-Tønnesen |
| 9 Behandling av de enkelte kreftsykdommer |
|
|
| 9.1 Leukemier, lymfomer og histiocytoser |
|
|
| 9.1 - Avsnitt "Leukemier" | Marit Hellebostad, Ann Elisabeth Åsberg og Bernward Zeller | Marit Hellebostad, Bernward Zeller |
| 9.1 - Avsnitt "Non Hodgkin lymfom" | Jochen Büchner | Maria Winther Gunnes |
| 9.1 - Avsnitt "Hodgkin lymfom" | Alexander Fosså | Alexander Fosså |
| 9.1 - Avsnitt "Histiocytoser" | Marit Hellebostad | Monica Cheng Munthe-Kaas, Finn Wesenberg |
| 9.2 Svulster i sentralnervesystemet |
|
|
| 9.2 - Avsnitt "Lavgradig gliom" | Tore Stokland | Tore Stokland |
| 9.2 - Avsnitt "Høygradige gliomer og Diffuse ponsgliomer" | Tore Stokland | Tore Stokland |
| 9.2 - Avsnitt "Ependymom" | Finn Wesenberg og Bernt J. Due-Tønnessen | Finn Wesenberg og Bernt J. Due-Tønnessen |
| 9.2 - Avsnitt "Medulloblastom" | Finn Wesenberg og Bernt J. Due-Tønnessen | Finn Wesenberg og Bernt J. Due-Tønnessen |
| 9.2 - Avsnitt "Supratentoriell PNET" | Finn Wesenberg og Bernt J. Due-Tønnessen | Finn Wesenberg og Bernt J. Due-Tønnessen |
| 9.2 - Avsnitt "Germinalcellesvulster i CNS" | Randi Nygaard | Randi Nygård, Petter Brandal |
| 9.2 - Avsnitt "Kraniofaryngeom" | Johan Cappelen og Tore Stokland | Tore Stokland |
| 9.2 - Avsnitt "Plexus Chorioideus-svulster" | Bernward Zeller | Bernward Zeller |
| 9.3 Solide svulster utenfor nervesystemet |
|
|
| 9.3 - Avsnitt "Sarkomer" | Heidi Glosli, Einar Stensvold og Øystein Drivenes | Heidi Glosli |
| 9.3 - Avsnitt "Nyresvulster" | Eva Widing | Eva Widing |
| 9.3 - Avsnitt "Nevroblastom" | Ellen Ruud, Kristin Bjørnland og Øystein Drivenes | Ellen Ruud, Kristin Bjørnland, Maria W. Gunnes |
| 9.3 - Avsnitt "Leversvulster" | Eva Widing og Kristin Bjørnland | Eva Widing |
| 9.3 - Avsnitt "Germinalcellesvulster utenfor CNS" | Eva Widing | Dorota Wojcik |
| 9.3 - Avsnitt "Retinoblastom (RB)" | Einar Stensvold og Randi Skarpaas Tranheim | Einar Stensvold og Randi Skarpaas Tranheim |
| 9.4 - Avsnitt "Sjeldne svulster" |
|
|
| 10 Støttebehandling og rettigheter |
|
|
| 10.1 Medisinsk støttebehandling | Ann Elisabeth Åsberg | Maria Winther Gunnes |
| 10.2 Psykososiale tiltak | Kjell Magnus Antonsen | Kjell Magnus Antonsen |
| 10.3 Rettigheter/støtteordninger | Sidsel Randklev | Sidsel Randklev |
| 10.4 Vaksinasjoner ved barnekreft | Trond Flægstad og Per Kristian Knudsen | Trond Flægstad, Per Kristian Knudsen, Margrete Greve-Isdahl |
| 10.5 Vaksinasjon etter allogen stamcelletransplantasjon til barn | Anders Glomstein | Jochen Büchner og Margrete Greve-Isdahl |
| 11 Oppfølging og etterkontroll. Senskader | Inga María Rinvoll Jóhannsdóttir, Bendik Lund, Adriani Kanellopoulos | Inga María Rinvoll Jóhannsdóttir, Bendik Lund, Adriani Kanellopoulos, Maria W. Gunnes |
| 11.1 Generelt |
|
|
| 11.2 Fysiske seneffekter |
|
|
| 11.3 Psykososiale seneffekter |
|
|
| 11.4 Oppfølging |
|
|
| 11.5 Nevropsykologisk oppfølging etter behandling for hjernesvulst | Torhild Berntsen | Torhild Berntsen |
| 12 Palliativ og terminal behandling | Marit Hellebostad og Thore Henrichsen | Marit Hellebostad |
| 13 Prosess og metode for utarbeiding av retningslinjene | Helsedirektoratet |
|
Foreliggende fjerde versjon av handlingsprogrammet - utgitt 26.05.2020 (IS‑2925)
Revisjonsgruppe 2019/2020
- Niklas Stabell (leder), Barneklinikken, Universitetssykehuset Nord Norge, Tromsø
- Bernt J. Due-Tønnessen, Nevrokirurgisk avdeling, Seksjon for barnenevrokirurgi Oslo universitetssykehus, Rikshospitalet
- Bernward Zeller, leder av kompetansesenteret for solide svulster hos barn, Oslo universitetssykehus Rikshospitalet
- Tove Nystad, Barneavdelingen, Universitetssykehuset Nord Norge, Tromsø
Revisjonsgruppen sendte januar 2020 revidert Nasjonalt handlingsprogram med retningslinjer for diagnostikk, behandling og oppfølging av barnekreft, til høring til lederne for:
- Faggruppen for svulster i sentralnervesystemet hos barn
- Faggruppen for solide svulster utenfor CNS hos barn
- Norsk barneleukemigruppe
I fjerde revisjon av Handlingsprogrammet er kapitelene om diagnostikk og behandling revidert, i likhet med kapitelet som omhandler medisinsk støttebehandling, rettigheter/støtteordninger og vaksinasjon ved barnekreft og etter allogen stamcelletransplantasjon.
I behandlingskapitelet er det lagt til et nytt kapitel om AT/RT svulstgruppen. Øvrige kapitel er uendret fra tredje versjon av Handlingsprogrammet.
I likhet med tidligere versjoner av Handlingsprogrammet er behandlingskapitelene for de ulike svulstgruppene og maligne blodsykdommene oppdatert med gjeldene nordiske og internasjonale behandlingsprotokoller, som faggruppene på nasjonalt nivå i enighet anbefaler. Behandlingsprotokollene er i de fleste tilfeller studier, enten kun registerstudier eller med intervensjon/utprøvende behandling. Studieprotokollene er godkjent av helsemyndighetene og i hver helseregion før pasientene kan inkluderes i behandlingsstudiene. I tilfeller der det ikke foreligger behandlingsstudier er det utarbeidet anbefalinger for behandling (« best available treatment»), eller at det vises til lovende forskning og behandlingsmuligheter, uten at det kan anses som etablerte behandlinger. I slike situasjoner, som ved behandlingsrefraktær sykdom, er det opp til hver behandlingsinstitusjon å søke kunnskap om hva som er den beste behandling for hver enkelt pasient, ofte i samarbeid med nasjonal og internasjonal ekspertise. Dersom behandlingen mangler godkjenning for bruk i Norge for respektive sykdom, må behandler søke lokal klinikk om gjennomføring av behandling og evt. helseforetaket («beslutingsforum»), ikke minst da behandlingene kan være kostbare. I noen tilfeller vil det også være mulighet for å søke om vederlagsfri behandling fra produsenten («compassionate use»).
Hovedforfattere av de enkelte kapitler
|
| Opprinnelig(e) forfatter(e) | Revisjon 2020 |
|---|---|---|
| 1 Innledning | Bernward Zeller og Finn Wesenberg | Uendret |
| 2 Epidemiologi | Bernward Zeller og Finn Wesenberg | Uendret |
| 3 Forebygging |
|
|
| 4 Forløpstider |
| Helsedirektoratet |
| 4.1 Om pakkeforløp for kreft |
|
|
| 4.2 Forløpstider for kreft hos barn |
|
|
| 5 Tidlig diagnostikk/screening |
|
|
| 6 Diagnostisering |
|
|
| 6.1 Symptomer, utredning, stadieinndeling | Bernward Zeller | Uendret |
| 6.2 Patologisk-anatomisk diagnostikk av barnekreft | Klaus Beiske | Klaus Beiske |
| 6.3 Integrert diagnostikk |
| Uendret |
| 6.4 Biobank | Wenche Reed | Monica Munthe-Kaas |
| 7 Genetikk |
|
|
| 7.1 Arv og kreft hos barn | Ketil Heimdal | Ketil Heimdal |
| 7.2 Molekylærgenetiske forhold | Ketil Heimdal | Ketil Heimdal |
| 8 Behandling |
|
|
| 8.1 Generelt om medikamentell behandling | Bernward Zeller | Uendret |
| 8.2 Kirurgi |
|
|
| 8.2 - Avsnitt "Kirurgisk behandling av solide svulster utenfor sentralnærvesystemet" | Kristin Bjørnland og Øystein Drivenes | Uendret |
| 8.2 - Avsnitt "Kirurgisk behandling av svulster i sentralnervestystemet" | Bernt J. Due-Tønnessen | Uendret |
| 8.3 Strålebehandling | Petter Brandal, Knut Lote og Odd Monge | Uendret |
| 8.4 Utprøvende behandling | Finn Wesenberg | Uendret |
| 8.5 Akutte onkologiske tilstander | Anne Elisabeth Åsberg | Uendret |
| 9 Behandling av de enkelte kreftsykdommer |
|
|
| 9.1 Leukemier, lymfomer og histiocytoser |
|
|
| 9.1 - Avsnitt "Leukemier" | Marit Hellebostad, Ann Elisabeth Åsberg og Bernward Zeller | Bernward Zeller, Inga María Jóhannsdóttir |
| 9.1 - Avsnitt "Non Hodgkin lymfom" | Jochen Büchner | Maria Winther Gunnes |
| 9.1 - Avsnitt "Hodgkin lymfom" | Alexander Fosså | Alexander Fosså |
| 9.1 - Avsnitt "Histiocytoser" | Marit Hellebostad | Monica Cheng Munthe-Kaas, Maria Winther Gunnes |
| 9.2 - Avsnitt "Svulster i sentralnervesystemet" |
|
|
| 9.2 - Avsnitt "Lavgradig gliom" | Tore Stokland | Tore Stokland |
| 9.2 - Avsnitt "Høygradige gliomer og Diffuse ponsgliomer" | Tore Stokland | Ingrid Torsvik |
| 9.2 - Avsnitt "Ependymom" | Finn Wesenberg og Bernt J. Due-Tønnessen | Ingrid Torsvik |
| 9.2 - Avsnitt "Medulloblastom" | Finn Wesenberg og Bernt J. Due-Tønnessen | Anne Vestli, Bernt J. Due-Tønnessen, Petter Brandal |
| 9.2 - Avsnitt "Andre embryonale hjernesvulster" | Finn Wesenberg og Bernt J. Due-Tønnessen | Anne Vestli, Bernt J. Due-Tønnessen, Petter Brandal |
| 9.2 - Avsnitt "Atypisk teratoid-rhabdoid tumor" |
| Anne Grete Bechensteen |
| 9.2 - AVsnitt "Germinalcellesvulster i CNS" | Randi Nygaard | Kristin Solem |
| 9.2 - Avsnitt "Kraniofaryngeom" | Johan Cappelen og Tore Stokland | Tore Stokland, Bernt J. Due-Tønnessen |
| 9.2 - Avsnitt "Plexus Chorioideus-svulster" | Bernward Zeller | Bernt J. Due-Tønnessen |
| 9.3 Solide svulster utenfor nervesystemet |
|
|
| 9.3 - Avsnitt "Sarkomer" | Heidi Glosli, Einar Stensvold og Øystein Drivenes | Heidi Glosli |
| 9.3 - Avsnitt "Nyresvulster" | Eva Widing | Marta Maria Burman |
| 9.3 - Avsnitt "Nevroblastom" | Ellen Ruud, Kristin Bjørnland og Øystein Drivenes | Ellen Ruud |
| 9.3 - Avsnitt "Leversvulster" | Eva Widing og Kristin Bjørnland | Marta Maria Burman |
| 9.3 - Avsnitt "Germinalcellesvulster utenfor CNS" | Eva Widing | Dorota Wojcik |
| 9.3 - Avsnitt "Retinoblastom (RB)" | Einar Stensvold og Randi Skarpaas Tranheim | Anne Vestli |
| 9.4 Sjeldne svulster |
| Uendret |
| 10 Støttebehandling og rettigheter |
|
|
| 10.1 Medisinsk støttebehandling | Ann Elisabeth Åsberg | Maria Winther Gunnes, Niklas Stabell |
| 10.2 Psykososiale tiltak | Kjell Magnus Antonsen | Kjell Magnus Antonsen |
| 10.3 Rettigheter/støtteordninger | Sidsel Randklev | Edel Grane |
| 10.4 Vaksinasjoner ved barnekreft | Trond Flægstad og Per Kristian Knudsen | Margrete Greve-Isdahl, Jochen Büchner |
| 10.5 Vaksinasjon etter allogen stamcelletransplantasjon til barn | Anders Glomstein | Margrete Greve-Isdahl, Jochen Büchner |
| 11 Oppfølging og etterkontroll. Senskader | Inga María Rinvoll Jóhannsdóttir, Bendik Lund, Adriani Kanellopoulos | Uendret |
| 11.1 Generelt |
|
|
| 11.2 Fysiske seneffekter |
|
|
| 11.3 Psykososiale seneffekter |
|
|
| 11.4 Oppfølging |
|
|
| 11.5 Nevropsykologisk oppfølging etter behandling for hjernesvulst | Torhild Berntsen | Uendret |
| 12 Palliativ og terminal behandling | Marit Hellebostad og Thore Henrichsen | Uendret |
| 13 Prosess og metode for utarbeiding av retningslinjene | Helsedirektoratet |
|
Habilitet
Sist faglig oppdatert: 26.05.2020
Arbeidsgruppenes medlemmer har i forbindelse med arbeidet bedt om å oppgi potensielle interessekonflikter. Helsedirektoratet har vurdert arbeidsgruppens medlemmer som habile i forhold til arbeid med dette nasjonale handlingsprogrammet.
Ressursmessige konsekvenser
Sist faglig oppdatert: 26.05.2020
De forslag som fremlegges ved disse retningslinjene vil ikke føre til økte kostnader for spesialisthelsetjenesten.
Oppdatering av retningslinjene
Sist faglig oppdatert: 26.05.2020
Utviklingen av ny behandling på kreftområdet går raskt. Handlingsprogrammet vil vurderes årlig og om nødvendig oppdateres.
Oppdateringen utføres av en arbeidsgruppe som består av fagpersoner oppnevnt av RHF-ene og Helsedirektoratet. Kunnskapssenteret i Folkehelseinstituttet skal også bidra i arbeid med oppdatering av handlingsprogrammet.
De oppdaterte retningslinjene vil publiseres på helsedirektoratet.no og helsebiblioteket.no (web-versjon).
Referanser
Sist faglig oppdatert: 26.05.2020
Abildgaard, L., Ellebaek, E., Gustafsson, G., Abrahamsson, J., Hovi, L., Jonmundsson, G., . . . Hasle, H. (2006). Optimal treatment intensity in children with Down syndrome and myeloid leukaemia: data from 56 children treated on NOPHO-AML protocols and a review of the literature. Ann Hematol, 85(5), 275-280.
Abla, O., Egeler, R. M., & Weitzman, S. (2010). Langerhans cell histiocytosis: Current concepts and treatments. Cancer Treat Rev, 36(4), 354-359.
Abramson, D. H., Greenfield, D. S., & Ellsworth, R. M. (1992). Bilateral retinoblastoma. Correlations between age at diagnosis and time course for new intraocular tumors. Ophthalmic Paediatr Genet, 13(1), 1-7.
Abzug, M. J. (2009). Vaccination in the immunocompromised child: a probe of immune reconstitution. Pediatr Infect Dis J, 28(3), 233-236.
Akuttveileder i pediatri [e-bok]. (2013). (3 utg.). Tromsø: Norsk barnelegeforening. Hentet fra http://www.helsebiblioteket.no/retningslinjer/akuttveileder-i-pediatri/forside
Andolina, J. R., Neudorf, S. M., & Corey, S. J. (2012). How I treat childhood CML. Blood, 119(8), 1821-1830.
Ansell, S. M., Lesokhin, A. M., Borrello, I., Halwani, A., Scott, E. C., Gutierrez, M., . . . Armand, P. (2015). PD-1 blockade with nivolumab in relapsed or refractory Hodgkin's lymphoma. N Engl J Med, 372(4), 311-319. https://doi.org/10.1056/NEJMoa1411087
Aronson, D. C., Schnater, J. M., Staalman, C. R., Weverling, G. J., Plaschkes, J., Perilongo, G., . . . Vos, A. (2005). Predictive value of the pretreatment extent of disease system in hepatoblastoma: results from the International Society of Pediatric Oncology Liver Tumor Study Group SIOPEL-1 study. J Clin Oncol, 23(6), 1245-1252.
Australian Technical Advisory Group on Immunisation (ATAGI). (2018). Australian Immunisation Handbook. Canberra: Australian Government Department of Health. Hentet fra https://immunisationhandbook.health.gov.au/
Bate, J., Borrow, R., Chisholm, J., Clark, S., Dixon, E., Faust, S. N., . . . Gray, J. C. (2019). 13-valent Pneumococcal Conjugate Vaccine in children with acute lymphoblastic leukaemia: protective immunity can be achieved on completion of treatment. Clinical infectious diseases : an official publication of the Infectious Diseases Society of America, Oct 5, [epub ahead of print]. https://doi.org/10.1093/cid/ciz965
Beckwith, J. B. (1998). Nephrogenic rests and the pathogenesis of Wilms tumor: developmental and clinical considerations. Am J Med Genet, 79(4), 268-273.
Bielack, S. S., Smeland, S., Whelan, J. S., Marina, N., Jovic, G., Hook, J. M., . . . Bernstein, M. (2015). Methotrexate, Doxorubicin, and Cisplatin (MAP) Plus Maintenance Pegylated Interferon Alfa-2b Versus MAP Alone in Patients With Resectable High-Grade Osteosarcoma and Good Histologic Response to Preoperative MAP: First Results of the EURAMOS-1 Good Response Randomized Controlled Trial. J Clin Oncol, 33(20), 2279-2287. https://doi.org/10.1200/jco.2014.60.0734
Biological Medicine for Diffuse Intrinsic Pontine Glioma (DIPG) Eradication (BIOMEDE). (2014-). [pågående studie]. (NCT02233049). Hentet fra https://clinicaltrials.gov/ct2/show/NCT02233049
Bisogno, G., & Bergeron, C. (2005). Soft tissue sarcoma. I: P. A. Voûte, A. Barrett, M. C. G. Stevens , & H. N. Carson (red.), Cancer in children: clinical management (s. 255-279). Oxford: Oxford University Press.
Bosetti, C., Bertuccio, P., Chatenoud, L., Negri, E., Levi, F., & La, V. C. (2010). Childhood cancer mortality in Europe, 1970-2007. Eur J Cancer, 46(2), 384-394.
Bouffet, E., Jakacki, R., Goldman, S., Hargrave, D., Hawkins, C., Shroff, M., . . . Baruchel, S. (2012). Phase II study of weekly vinblastine in recurrent or refractory pediatric low-grade glioma. J Clin Oncol, 30(12), 1358-1363.
Bouffet, E., Tabori, U., Huang, A., & Bartels, U. (2009). Ependymoma: lessons from the past, prospects for the future. Childs Nerv Syst, 25(11), 1383-1384; author reply 1385. https://doi.org/10.1007/s00381-009-0915-6
Braunstein, S., Raleigh, D., Bindra, R., Mueller, S., & Haas-Kogan, D. (2017). Pediatric high-grade glioma: current molecular landscape and therapeutic approaches. J Neurooncol, 134(3), 541-549. https://doi.org/10.1007/s11060-017-2393-0
Cage, T. A., Clark, A. J., Aranda, D., Gupta, N., Sun, P. P., Parsa, A. T., & Auguste, K. I. (2013). A systematic review of treatment outcomes in pediatric patients with intracranial ependymomas. J Neurosurg Pediatr, 11(6), 673-681. https://doi.org/10.3171/2013.2.peds12345
Cancer in children: clinical management. (2005). (5 utg.). Oxford: Oxford University Press.
Caniza, M. A., Hunger, S. P., Schrauder, A., Valsecchi, M. G., Pui, C. H., & Masera, G. (2012). The controversy of varicella vaccination in children with acute lymphoblastic leukemia. Pediatr Blood Cancer, 58(1), 12-16. https://doi.org/10.1002/pbc.22759
Carreras, E., Dufour, C., Mohty, M., & Kröger, N. (red.). (2019). The EBMT Handbook: Hematopoietic Stem Cell Transplantation and Cellular Therapies. Cham, Switzerland: Springer. Hentet fra https://pubmed.ncbi.nlm.nih.gov/32091673/
Ceppi, F., Weitzman, S., Woessmann, W., Davies, K., Lassaletta, A., Reismuller, B., . . . Alexander, S. (2016). Safety and efficacy of intrathecal rituximab in children with B cell lymphoid CD20+ malignancies: An international retrospective study. American journal of hematology, 91(5), 486-491. https://doi.org/10.1002/ajh.24329
Childhood cancer in the Nordic countries. (2008). Linköping: Nordic society of Pediatric Haematology and Oncology.
Cohen, B. H., Zeltzer, P. M., Boyett, J. M., Geyer, J. R., Allen, J. C., Finlay, J. L., . . . et al. (1995). Prognostic factors and treatment results for supratentorial primitive neuroectodermal tumors in children using radiation and chemotherapy: a Childrens Cancer Group randomized trial. J Clin Oncol, 13(7), 1687-1696. https://doi.org/10.1200/jco.1995.13.7.1687
Cohen, K. J., Pollack, I. F., Zhou, T., Buxton, A., Holmes, E. J., Burger, P. C., . . . Heideman, R. L. (2011). Temozolomide in the treatment of high-grade gliomas in children: a report from the Children's Oncology Group. Neuro Oncol, 13(3), 317-323. https://doi.org/10.1093/neuonc/noq191
Cohn, S. L., Pearson, A. D., London, W. B., Monclair, T., Ambros, P. F., Brodeur, G. M., . . . Matthay, K. K. (2009). The International Neuroblastoma Risk Group (INRG) classification system: an INRG Task Force report. J Clin Oncol, 27(2), 289-297.
Cook, R. L., O'Dwyer, N. J., Parker, H. M., Donges, C. E., Cheng, H. L., Steinbeck, K. S., . . . O'Connor, H. T. (2017). Iron Deficiency Anemia, Not Iron Deficiency, Is Associated with Reduced Attention in Healthy Young Women. Nutrients, 9(11), 1216. https://doi.org/10.3390/nu9111216
Cordonnier, C., Labopin, M., Chesnel, V., Ribaud, P., De La Camara, R., Martino, R., . . . Ljungman, P. (2009). Randomized study of early versus late immunization with pneumococcal conjugate vaccine after allogeneic stem cell transplantation. Clinical infectious diseases : an official publication of the Infectious Diseases Society of America, 48(10), 1392-1401. https://doi.org/10.1086/598324
Creutzig, U., Reinhardt, D., Diekamp, S., Dworzak, M., Stary, J., & Zimmermann, M. (2005). AML patients with Down syndrome have a high cure rate with AML-BFM therapy with reduced dose intensity. Leukemia, 19(8), 1355-1360.
Dickerman, J. D. (2007). The late effects of childhood cancer therapy. Pediatrics, 119(3), 554-568.
Dimaras, H., Kimani, K., Dimba, E. A., Gronsdahl, P., White, A., Chan, H. S., & Gallie, B. L. (2012). Retinoblastoma. Lancet, 379(9824), 1436-1446. https://doi.org/10.1016/s0140-6736(11)61137-9
Dorffel, W., Ruhl, U., Luders, H., Claviez, A., Albrecht, M., Bokkerink, J., . . . Schellong, G. (2013). Treatment of children and adolescents with Hodgkin lymphoma without radiotherapy for patients in complete remission after chemotherapy: final results of the multinational trial GPOH-HD95. J Clin Oncol, 31(12), 1562-1568.
Due-Tønnessen, B. J., Helseth, E., Scheie, D., Skullerud, K., Aamodt, G., & Lundar, T. (2002). Long-term outcome after resection of benign cerebellar astrocytomas in children and young adults (0-19 years): report of 110 consecutive cases. Pediatr Neurosurg, 37(2), 71-80. https://doi.org/10.1159/000065108
Due-Tønnessen, B. J., Lundar, T., Egge, A., & Scheie, D. (2013). Neurosurgical treatment of low-grade cerebellar astrocytoma in children and adolescents: a single consecutive institutional series of 100 patients. J Neurosurg Pediatr, 11(3), 245-249.
Ellison, D. W., Kocak, M., Figarella-Branger, D., Felice, G., Catherine, G., Pietsch, T., . . . Grundy, R. G. (2011). Histopathological grading of pediatric ependymoma: reproducibility and clinical relevance in European trial cohorts. Journal of negative results in biomedicine, 10, 7. https://doi.org/10.1186/1477-5751-10-7
Esposito, S., Cecinati, V., Brescia, L., & Principi, N. (2010). Vaccinations in children with cancer. Vaccine, 28(19), 3278-3284.
Foreldrekontakttjenesten. (2013). [nettside]. Oslo: Barnekreftforeningen. Hentet 8. aug. 2013, fra http://www.barnekreftforeningen.no/for-medlemmer/foreldre-m/
Forestier, E., & Schmiegelow, K. (2006). The incidence peaks of the childhood acute leukemias reflect specific cytogenetic aberrations. J Pediatr Hematol Oncol, 28(8), 486-495.
Fossey, M., Li, H., Afzal, S., Carret, A. S., Eisenstat, D. D., Fleming, A., . . . Lafay-Cousin, L. (2017). Atypical teratoid rhabdoid tumor in the first year of life: the Canadian ATRT registry experience and review of the literature. J Neurooncol, 132(1), 155-162. https://doi.org/10.1007/s11060-016-2353-0
Fouladi, M., Hunt, D. L., Pollack, I. F., Dueckers, G., Burger, P. C., Becker, L. E., . . . Finlay, J. L. (2003). Outcome of children with centrally reviewed low-grade gliomas treated with chemotherapy with or without radiotherapy on Children's Cancer Group high-grade glioma study CCG-945. Cancer, 98(6), 1243-1252. https://doi.org/10.1002/cncr.11637
Friend, S. H., Bernards, R., Rogelj, S., Weinberg, R. A., Rapaport, J. M., Albert, D. M., & Dryja, T. P. (1986). A human DNA segment with properties of the gene that predisposes to retinoblastoma and osteosarcoma. Nature, 323(6089), 643-646.
Gallamini, A., Hutchings, M., Rigacci, L., Specht, L., Merli, F., Hansen, M., . . . Levis, A. (2007). Early interim 2-[18F]fluoro-2-deoxy-D-glucose positron emission tomography is prognostically superior to international prognostic score in advanced-stage Hodgkin's lymphoma: a report from a joint Italian-Danish study. J Clin Oncol, 25(24), 3746-3752. https://doi.org/10.1200/jco.2007.11.6525
Gatta, G., Botta, L., Rossi, S., Aareleid, T., Bielska-Lasota, M., Clavel, J., . . . Peris-Bonet, R. (2014). Childhood cancer survival in Europe 1999-2007: results of EUROCARE-5--a population-based study. The Lancet. Oncology, 15(1), 35-47. https://doi.org/10.1016/s1470-2045(13)70548-5
Gatta, G., Capocaccia, R., Stiller, C., Kaatsch, P., Berrino, F., & Terenziani, M. (2005). Childhood cancer survival trends in Europe: a EUROCARE Working Group study. J Clin Oncol, 23(16), 3742-3751.
Gatta, G., Zigon, G., Capocaccia, R., Coebergh, J. W., Desandes, E., Kaatsch, P., . . . Stiller, C. A. (2009). Survival of European children and young adults with cancer diagnosed 1995-2002. Eur J Cancer, 45(6), 992-1005.
Gaze, M. N., Chang, Y. C., Flux, G. D., Mairs, R. J., Saran, F. H., & Meller, S. T. (2005). Feasibility of dosimetry-based high-dose 131I-meta-iodobenzylguanidine with topotecan as a radiosensitizer in children with metastatic neuroblastoma. Cancer biotherapy & radiopharmaceuticals, 20(2), 195-199. https://doi.org/10.1089/cbr.2005.20.195
Generell veileder i pediatri [e-bok]. (2012). (2 utg.). Tromsø: Norsk barnelegeforening. Hentet fra http://www.helsebiblioteket.no/retningslinjer/pediatri/forside
Gianno F, A. M., Ferretti E, Massimino M, Arcella A, Giangaspero F. (2018). Pediatric high-grade glioma: A heterogeneous group of neoplasms with different molecular drivers. Glioma, (1), 117-124.
Gibson, B. E., Todd, A., Roberts, I., Pamphilon, D., Rodeck, C., Bolton-Maggs, P., . . . Turner, G. (2004). Transfusion guidelines for neonates and older children. Br J Haematol, 124(4), 433-453.
Gielen, G. H., Gessi, M., Buttarelli, F. R., Baldi, C., Hammes, J., zur Muehlen, A., . . . Pietsch, T. (2015). Genetic Analysis of Diffuse High-Grade Astrocytomas in Infancy Defines a Novel Molecular Entity. Brain pathology (Zurich, Switzerland), 25(4), 409-417. https://doi.org/10.1111/bpa.12210
Gnekow, A. K., Kandels, D., van Tilburg, C., Azizi, A. A., Opocher, E., Stokland, T., . . . Witt, O. (2019). SIOP-E-BTG and GPOH Guidelines for Diagnosis and Treatment of Children and Adolescents with Low Grade Glioma. Klinische Padiatrie, 231(3), 107-135. https://doi.org/10.1055/a-0889-8256
Gnekow, A. K., Packer, R., & Kortmann, R. D. (2004). Astrocytic tumors, low-grade. I: D. V. Walker, G. Perilongo, J. A. G. Punt, & R. Taylor (red.), Brain and spinal tumors of childhood (s. 245-276). London: Arnold.
Gobel, U., Calaminus, G., Engert, J., Kaatsch, P., Gadner, H., Bokkerink, J. P., . . . Harms, D. (1998). Teratomas in infancy and childhood. Med Pediatr Oncol, 31(1), 8-15.
Gobel, U., Schneider, D. T., Calaminus, G., Haas, R. J., Schmidt, P., & Harms, D. (2000). Germ-cell tumors in childhood and adolescence. GPOH MAKEI and the MAHO study groups. Ann Oncol, 11(3), 263-271.
Goldman, S., Smith, L., Anderson, J. R., Perkins, S., Harrison, L., Geyer, M. B., . . . Cairo, M. S. (2013). Rituximab and FAB/LMB 96 chemotherapy in children with Stage III/IV B-cell non-Hodgkin lymphoma: a Children's Oncology Group report. Leukemia, 27(5), 1174-1177.
Gregersen, P. A., Urbak, S. F., Funding, M., Overgaard, J., Jensen, U. B., & Alsner, J. (2016). Danish retinoblastoma patients 1943-2013 - genetic testing and clinical implications. Acta Oncol, 55(4), 412-417. https://doi.org/10.3109/0284186X.2015.1099732
Grill, J., Le Deley, M. C., Gambarelli, D., Raquin, M. A., Couanet, D., Pierre-Kahn, A., . . . Kalifa, C. (2001). Postoperative chemotherapy without irradiation for ependymoma in children under 5 years of age: a multicenter trial of the French Society of Pediatric Oncology. J Clin Oncol, 19(5), 1288-1296. https://doi.org/10.1200/jco.2001.19.5.1288
Grundy, R. G., Wilne, S. A., Weston, C. L., Robinson, K., Lashford, L. S., Ironside, J., . . . Machin, D. (2007). Primary postoperative chemotherapy without radiotherapy for intracranial ependymoma in children: the UKCCSG/SIOP prospective study. The Lancet. Oncology, 8(8), 696-705. https://doi.org/10.1016/s1470-2045(07)70208-5
Gunnes, M. W., Lie, R. T., Bjørge, T., Ghaderi, S., Ruud, E., Syse, A., & Moster, D. (2016). Reproduction and marriage among male survivors of cancer in childhood, adolescence and young adulthood: a national cohort study. British journal of cancer, 114(3), 348-356. https://doi.org/10.1038/bjc.2015.455
Gupta, A., & Dwivedi, T. (2017). A Simplified Overview of World Health Organization Classification Update of Central Nervous System Tumors 2016. Journal of neurosciences in rural practice, 8(4), 629-641. https://doi.org/10.4103/jnrp.jnrp_168_17
Gustafsson, B., Hellebostad, M., Ifversen, M., Sander, B., & Hasle, H. (2011). Acute respiratory failure in 3 children with juvenile myelomonocytic leukemia. J Pediatr Hematol Oncol, 33(8), e363-e367.
Gustafsson, G., Schmiegelow, K., Forestier, E., Clausen, N., Glomstein, A., Jonmundsson, G., . . . Saarinen-Pihkala, U. M. (2000). Improving outcome through two decades in childhood ALL in the Nordic countries: the impact of high-dose methotrexate in the reduction of CNS irradiation. Nordic Society of Pediatric Haematology and Oncology (NOPHO). Leukemia, 14(12), 2267-2275.
Hagberg-van't Hooft, I. (2005). Cognitive rehabilitation in children with acquired brain injuries [doktorgrad]. Stockholm: Karolinska institutet, Department of Women's and Children's Health. Hentet fra http://publications.ki.se/xmlui/bitstream/handle/10616/39503/thesis.pdf?sequence=1
Hamre, H., Zeller, B., Kanellopoulos, A., Kiserud, C. E., Aakhus, S., Lund, M. B., . . . Ruud, E. (2012). High Prevalence of Chronic Fatigue in Adult Long-Term Survivors of Acute Lymphoblastic Leukemia and Lymphoma During Childhood and Adolescence. Journal of Adolescent and Young Adult Oncology, 2(1), 2-9. https://doi.org/10.1089/jayao.2012.0015
Hargrave, D. (2009). Paediatric high and low grade glioma: the impact of tumour biology on current and future therapy. Br J Neurosurg, 23(4), 351-363.
Hasle, H., Abrahamsson, J., Forestier, E., Ha, S. Y., Heldrup, J., Jahnukainen, K., . . . Zeller, B. (2012). Gemtuzumab ozogamicin as postconsolidation therapy does not prevent relapse in children with AML: results from NOPHO-AML 2004. Blood, 120(5), 978-984.
Helsedirektoratet. (2013). Beslutningsprosesser for begrensning av livsforlengende behandling hos alvorlige syke og døende (rev. utg.). (IS-2091). Oslo: Helsedirektoratet. Hentet fra https://www.helsedirektoratet.no/veiledere/beslutningsprosesser-ved-begrensning-av-livsforlengende-behandling
Helsedirektoratet. (4. mai 2017). Palliasjon til barn og unge: nasjonal faglig retningslinje. [nettdokument]. Oslo: Helsedirektoratet. Hentet 5. mai 2020, fra https://www.helsedirektoratet.no/retningslinjer/palliasjon-til-barn-og-unge
Helsedirektoratet. (2018). Nasjonalt handlingsprogram med retningslinjer for diagnostikk, behandling og oppfølging av sarkom (3. utg.). (IS-2697). Oslo: Helsedirektoratet. Hentet fra https://www.helsedirektoratet.no/retningslinjer/sarkomer-handlingsprogram
Helsedirektoratet. (2019). Nasjonalt handlingsprogram med retningslinjer for diagnostikk, behandling og oppfølging av maligne lymfomer (IS-2747). Oslo: Helsedirektoratet. Hentet fra https://www.helsedirektoratet.no/retningslinjer/lymfekreft-handlingsprogram
Henter, J. I., Elinder, G., Soder, O., & Ost, A. (1991). Incidence in Sweden and clinical features of familial hemophagocytic lymphohistiocytosis. Acta Paediatr Scand, 80(4), 428-435.
Henter, J. I., Horne, A., Arico, M., Egeler, R. M., Filipovich, A. H., Imashuku, S., . . . Janka, G. (2007). HLH-2004: Diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer, 48(2), 124-131.
Hodgson, S. V., & Maher, E. R. (1999). A practical guide to human cancer genetics (2 utg.). Cambridge: Cambridge University Press.
Hwang, E. I., Jakacki, R. I., Fisher, M. J., Kilburn, L. B., Horn, M., Vezina, G., . . . Packer, R. J. (2013). Long-term efficacy and toxicity of bevacizumab-based therapy in children with recurrent low-grade gliomas. Pediatr Blood Cancer, 60(5), 776-782.
An International Clinical Program for the Diagnosis and Treatment of Children With Ependymoma (SIOP-EP-II). (2015-2026). [pågående studie]. (NCT02265770). Hentet fra https://clinicaltrials.gov/ct2/show/NCT02265770
Jakacki, R. I., Cohen, K. J., Buxton, A., Krailo, M. D., Burger, P. C., Rosenblum, M. K., . . . Pollack, I. F. (2016). Phase 2 study of concurrent radiotherapy and temozolomide followed by temozolomide and lomustine in the treatment of children with high-grade glioma: a report of the Children's Oncology Group ACNS0423 study. Neuro Oncol, 18(10), 1442-1450. https://doi.org/10.1093/neuonc/now038
Janka, G. E. (2007). Familial and acquired hemophagocytic lymphohistiocytosis. Eur J Pediatr, 166(2), 95-109.
Japp, A. S., Klein-Hitpass, L., Denkhaus, D., & Pietsch, T. (2017). OTX2 Defines a Subgroup of Atypical Teratoid Rhabdoid Tumors With Close Relationship to Choroid Plexus Tumors. Journal of neuropathology and experimental neurology, 76(1), 32-38. https://doi.org/10.1093/jnen/nlw101
Johann, P. D., Erkek, S., Zapatka, M., Kerl, K., Buchhalter, I., Hovestadt, V., . . . Kool, M. (2016). Atypical Teratoid/Rhabdoid Tumors Are Comprised of Three Epigenetic Subgroups with Distinct Enhancer Landscapes. Cancer Cell, 29(3), 379-393. https://doi.org/10.1016/j.ccell.2016.02.001
Kager, L., Zoubek, A., Potschger, U., Kastner, U., Flege, S., Kempf-Bielack, B., . . . Bielack, S. S. (2003). Primary metastatic osteosarcoma: presentation and outcome of patients treated on neoadjuvant Cooperative Osteosarcoma Study Group protocols. J Clin Oncol, 21(10), 2011-2018.
Kaste, S. C., Dome, J. S., Babyn, P. S., Graf, N. M., Grundy, P., Godzinski, J., . . . Jenkinson, H. (2008). Wilms tumour: prognostic factors, staging, therapy and late effects. Pediatr Radiol, 38(1), 2-17.
Khatua, S., Ramaswamy, V., & Bouffet, E. (2017). Current therapy and the evolving molecular landscape of paediatric ependymoma. Eur J Cancer, 70, 34-41. https://doi.org/10.1016/j.ejca.2016.10.013
Kim, J. W., Abramson, D. H., & Dunkel, I. J. (2007). Current management strategies for intraocular retinoblastoma. Drugs, 67(15), 2173-2185.
Kleinerman, R. A., Tucker, M. A., Tarone, R. E., Abramson, D. H., Seddon, J. M., Stovall, M., . . . Fraumeni, J. F., Jr. (2005). Risk of new cancers after radiotherapy in long-term survivors of retinoblastoma: an extended follow-up. J Clin Oncol, 23(10), 2272-2279. https://doi.org/10.1200/JCO.2005.05.054
Kleinerman, R. A., Yu, C. L., Little, M. P., Li, Y., Abramson, D., Seddon, J., & Tucker, M. A. (2012). Variation of second cancer risk by family history of retinoblastoma among long-term survivors. J Clin Oncol, 30(9), 950-957. https://doi.org/10.1200/JCO.2011.37.0239
Kohno, S., Kitajima, S., Sasaki, N., & Takahashi, C. (2016). Retinoblastoma tumor suppressor functions shared by stem cell and cancer cell strategies. World journal of stem cells, 8(4), 170-184. https://doi.org/10.4252/wjsc.v8.i4.170
Kolmannskog, S., Flaegstad, T., Helgestad, J., Hellebostad, M., Zeller, B., & Glomstein, A. (2007). Akutt lymfatisk leukemi hos barn i Norge 1992-2000. Tidsskr Nor Laegeforen, 127(11), 1493-1495.
Kreftoverlevere: ny kunnskap og nye muligheter i et langtidsperspektiv. (2009). Oslo: Gyldendal akademisk.
Krepischi, A. C., Capelli, L. P., Silva, A. G., de Araujo, E. S., Pearson, P. L., Heck, B., . . . Rosenberg, C. (2014). Large germline copy number variations as predisposing factor in childhood neoplasms. Future oncology (London, England), 10(9), 1627-1633. https://doi.org/10.2217/fon.14.41
Kaatsch, P., Steliarova-Foucher, E., Crocetti, E., Magnani, C., Spix, C., & Zambon, P. (2006). Time trends of cancer incidence in European children (1978-1997): report from the Automated Childhood Cancer Information System project. Eur J Cancer, 42(13), 1961-1971.
Larsen, I. K. (red.). (2018). Cancer in Norway 2018: Cancer incidence, mortality, survival and prevalence in Norway. Oslo: Cancer Registry of Norway. Hentet fra https://www.kreftregisteret.no/globalassets/cancer-in-norway/2018/cin2018.pdf
Late effects of childhood cancer. (2004). London: Arnold.
Levisohn, L., Cronin-Golomb, A., & Schmahmann, J. D. (2000). Neuropsychological consequences of cerebellar tumour resection in children: cerebellar cognitive affective syndrome in a paediatric population. Brain, 123 ( Pt 5), 1041-1050.
Lie, S. O., Abrahamsson, J., Clausen, N., Forestier, E., Hasle, H., Hovi, L., . . . Gustafsson, G. (2005). Long-term results in children with AML: NOPHO-AML Study Group--report of three consecutive trials. Leukemia, 19(12), 2090-2100.
Ljungman, P., Cordonnier, C., Einsele, H., Englund, J., Machado, C. M., Storek, J., & Small, T. (2009). Vaccination of hematopoietic cell transplant recipients. Bone marrow transplantation, 44(8), 521-526. https://doi.org/10.1038/bmt.2009.263
Lote, K., Hatlevoll, R., Bakke, S. J., Due-Tønnessen, B., Helseth, E., Lundar, T., . . . Skullerud, K. (2000). Langtids overlevelse etter kirurgi og strålebehandling av hjernesvulster i barnealder. Tidsskr Nor Laegeforen, 120(10), 1142-1145.
Louis, D. N., Ohgaki, H., Wiestler, O. D., Cavenee, W. K., Burger, P. C., Jouvet, A., . . . Kleihues, P. (2007). The 2007 WHO classification of tumours of the central nervous system. Acta neuropathologica, 114(2), 97-109. https://doi.org/10.1007/s00401-007-0243-4
Louis, D. N., Perry, A., Reifenberger, G., von Deimling, A., Figarella-Branger, D., Cavenee, W. K., . . . Ellison, D. W. (2016). The 2016 World Health Organization Classification of Tumors of the Central Nervous System: a summary. Acta neuropathologica, 131(6), 803-820. https://doi.org/10.1007/s00401-016-1545-1
Lu, V. M., Welby, J. P., Mahajan, A., Laack, N. N., & Daniels, D. J. (2019). Reirradiation for diffuse intrinsic pontine glioma: a systematic review and meta-analysis. Childs Nerv Syst, 35(5), 739-746. https://doi.org/10.1007/s00381-019-04118-y
Lundar, T., Due-Tonnessen, B. J., Scheie, D., & Brandal, P. (2014). Pediatric spinal ependymomas: an unpredictable and puzzling disease. Long-term follow-up of a single consecutive institutional series of ten patients. Childs Nerv Syst, 30(12), 2083-2088. https://doi.org/10.1007/s00381-014-2491-7
Lundar, T., Due-Tønnessen, B. J., Egge, A., Scheie, D., Brandal, P., Stensvold, E., & Due-Tønnessen, P. (2014). Neurosurgical treatment of pediatric low-grade midbrain tumors: a single consecutive institutional series of 15 patients. J Neurosurg Pediatr, 14(6), 598-603. https://doi.org/10.3171/2014.9.Peds1462
Mann, G., Attarbaschi, A., Schrappe, M., De, L. P., Peters, C., Hann, I., . . . Pieters, R. (2010). Improved outcome with hematopoietic stem cell transplantation in a poor prognostic subgroup of infants with mixed-lineage-leukemia (MLL)-rearranged acute lymphoblastic leukemia: results from the Interfant-99 Study. Blood, 116(15), 2644-2650.
Mann, J. R., Raafat, F., Robinson, K., Imeson, J., Gornall, P., Sokal, M., . . . Oakhill, A. (2000). The United Kingdom Children's Cancer Study Group's second germ cell tumor study: carboplatin, etoposide, and bleomycin are effective treatment for children with malignant extracranial germ cell tumors, with acceptable toxicity. J Clin Oncol, 18(22), 3809-3818.
Marin, M., Leung, J., & Gershon, A. A. (2019). Transmission of Vaccine-Strain Varicella-Zoster Virus: A Systematic Review. Pediatrics, 144(3). https://doi.org/10.1542/peds.2019-1305
Massimino, M., Gandola, L., Giangaspero, F., Sandri, A., Valagussa, P., Perilongo, G., . . . Madon, E. (2004). Hyperfractionated radiotherapy and chemotherapy for childhood ependymoma: final results of the first prospective AIEOP (Associazione Italiana di Ematologia-Oncologia Pediatrica) study. Int J Radiat Oncol Biol Phys, 58(5), 1336-1345. https://doi.org/10.1016/j.ijrobp.2003.08.030
Massimino, M., Gandola, L., Spreafico, F., Luksch, R., Collini, P., Giangaspero, F., . . . Fossati-Bellani, F. (2006). Supratentorial primitive neuroectodermal tumors (S-PNET) in children: A prospective experience with adjuvant intensive chemotherapy and hyperfractionated accelerated radiotherapy. Int J Radiat Oncol Biol Phys, 64(4), 1031-1037.
Mauz-Korholz, C., Gorde-Grosjean, S., Hasenclever, D., Shankar, A., Dorffel, W., Wallace, W. H., . . . Landman-Parker, J. (2007). Resection alone in 58 children with limited stage, lymphocyte-predominant Hodgkin lymphoma-experience from the European network group on pediatric Hodgkin lymphoma. Cancer, 110(1), 179-185.
Mauz-Korholz, C., Hasenclever, D., Dorffel, W., Ruschke, K., Pelz, T., Voigt, A., . . . Korholz, D. (2010). Procarbazine-free OEPA-COPDAC chemotherapy in boys and standard OPPA-COPP in girls have comparable effectiveness in pediatric Hodgkin's lymphoma: the GPOH-HD-2002 study. J Clin Oncol, 28(23), 3680-3686. https://doi.org/10.1200/jco.2009.26.9381
Medikamentell kreftbehandling: cytostatikaboken. (2009). (7 utg.). Oslo: Farmakologisk institutt, Det medisinske fakultet, Universitetet i Oslo.
Meinhardt, A., Burkhardt, B., Zimmermann, M., Borkhardt, A., Kontny, U., Klingebiel, T., . . . Reiter, A. (2010). Phase II window study on rituximab in newly diagnosed pediatric mature B-cell non-Hodgkin's lymphoma and Burkitt leukemia. J Clin Oncol, 28(19), 3115-3121.
Merchant, T. E., Li, C., Xiong, X., Kun, L. E., Boop, F. A., & Sanford, R. A. (2009). Conformal radiotherapy after surgery for paediatric ependymoma: a prospective study. The Lancet. Oncology, 10(3), 258-266. https://doi.org/10.1016/s1470-2045(08)70342-5
Minard-Colin, V., Brugieres, L., Reiter, A., Cairo, M. S., Gross, T. G., Woessmann, W., . . . Patte, C. (2015). Non-Hodgkin Lymphoma in Children and Adolescents: Progress Through Effective Collaboration, Current Knowledge, and Challenges Ahead. J Clin Oncol, 33(27), 2963-2974. https://doi.org/10.1200/jco.2014.59.5827
Monclair, T., Brodeur, G. M., Ambros, P. F., Brisse, H. J., Cecchetto, G., Holmes, K., . . . Pearson, A. D. (2009). The International Neuroblastoma Risk Group (INRG) staging system: an INRG Task Force report. J Clin Oncol, 27(2), 298-303. https://doi.org/10.1200/jco.2008.16.6876
Moskowitz, C. H., Walewski, J., Nademanee, A., Masszi, T., Agura, E., Holowiecki, J., . . . Sweetenham, J. (2018). Five-year PFS from the AETHERA trial of brentuximab vedotin for Hodgkin lymphoma at high risk of progression or relapse. Blood, 132(25), 2639-2642. https://doi.org/10.1182/blood-2018-07-861641
Mulhern, R. K. (2003). Cognitive sequelae. Brain Injury, 17(Suppl 1), 5.
Mulhern, R. K., & Butler, R. W. (2004). Neurocognitive sequelae of childhood cancers and their treatment. Pediatr Radiol, 7(1), 1-14.
Muller, H. L. (2010). Childhood craniopharyngioma: current controversies on management in diagnostics, treatment and follow-up. Expert Rev Neurother, 10(4), 515-524.
Murphree, A. L. (2005). Intraocular retinoblastoma: the case for a new group classification. Ophthalamic Oncology, Ophthalmology Clinics of North America, 18(1), 41-53.
Nasjonal helseplan (2007-2010). (2006). (Særtrykk av St.prp. nr. 1 (2006-2007) kapittel 6). Oslo: Helse- og omsorgsdepartemenetet. Hentet fra http://www.regjeringen.no/upload/HOD/Sykehus/Nasjonal_helseplan_Sartrykk.pdf
Nasjonalt kvalitetsregister for barnekreft. (2015). Årsrapport 2014. Oslo: Kreftregisteret. Hentet fra https://www.kreftregisteret.no/globalassets/publikasjoner-og-rapporter/arsrapporter/publisert-2015/aarsrapport_2014_barnekreft.pdf
Nasjonalt kvalitetsregister for barnekreft. (2018). Årsrapport 2018: resultater og forbedringstiltak fra Nasjonalt kvalitetsregister for barnekreft. Oslo: Kreftregisteret. Hentet fra https://www.kreftregisteret.no/globalassets/publikasjoner-og-rapporter/arsrapporter/publisert-2019/arsrapport-2018-barnekreft.pdf
Nordiske retningslinjer for retinoblastom. (2016). Oslo: Nordisk retinoblastom gruppe.
Oeffinger, K. C., Mertens, A. C., Sklar, C. A., Kawashima, T., Hudson, M. M., Meadows, A. T., . . . Robison, L. L. (2006). Chronic health conditions in adult survivors of childhood cancer. N Engl J Med, 355(15), 1572-1582.
Offit, K. (1998). Clinical cancer genetics: risk counselling and management. New York: Wiley-Liss.
Olsen, D. R., Bruland, O. S., Frykholm, G., & Norderhaug, I. N. (2007). Proton therapy - a systematic review of clinical effectiveness. Radiother Oncol, 83(2), 123-132.
Oppfølging av kreftoverlevere med særlig fokus på seneffekter: innstilling fra en arbeidsgruppe nedsatt av Helsedirektoratet. (2010). Oslo: Arbeidsgruppen. Hentet fra https://www.legeforeningen.no/contentassets/67b4a31a81c1491d933ecfee02332269/seneffekter-av-kreftbehandling-hoeringsvedlegg.pdf
Packer, R. J., Pfister, S., Bouffet, E., Avery, R., Bandopadhayay, P., Bornhorst, M., . . . Kieran, M. (2017). Pediatric low-grade gliomas: implications of the biologic era. Neuro Oncol, 19(6), 750-761. https://doi.org/10.1093/neuonc/now209
Pajtler, K. W., Mack, S. C., Ramaswamy, V., Smith, C. A., Witt, H., Smith, A., . . . Taylor, M. D. (2017). The current consensus on the clinical management of intracranial ependymoma and its distinct molecular variants. Acta neuropathologica, 133(1), 5-12. https://doi.org/10.1007/s00401-016-1643-0
Pajtler, K. W., Witt, H., Sill, M., Jones, D. T., Hovestadt, V., Kratochwil, F., . . . Pfister, S. M. (2015). Molecular Classification of Ependymal Tumors across All CNS Compartments, Histopathological Grades, and Age Groups. Cancer Cell, 27(5), 728-743. https://doi.org/10.1016/j.ccell.2015.04.002
Paulussen, M., Kovar, H., & Jürgens, H. (2005). Ewing sarcoma and peripheral primitive neuroectodermal tumour. I: P. A. Voûte, A. Barrett, M. C. G. Stevens, & H. N. Carson (red.), Cancer in children: clinical management (5 utg., s. 301-320). Oxford: Oxford University Press.
Pejavar, S., Polley, M. Y., Rosenberg-Wohl, S., Chennupati, S., Prados, M. D., Berger, M. S., . . . Haas-Kogan, D. (2012). Pediatric intracranial ependymoma: the roles of surgery, radiation and chemotherapy. J Neurooncol, 106(2), 367-375. https://doi.org/10.1007/s11060-011-0671-9
Perilongo, G. (2005). Considerations on the role of chemotherapy and modern radiotherapy in the treatment of childhood low grade glioma. J Neurooncol, 75(3), 301-307.
Perilongo, G., Maibach, R., Shafford, E., Brugieres, L., Brock, P., Morland, B., . . . Czauderna, P. (2009). Cisplatin versus cisplatin plus doxorubicin for standard-risk hepatoblastoma. N Engl J Med, 361(17), 1662-1670. https://doi.org/10.1056/NEJMoa0810613
Perilongo, G., Shafford, E., Maibach, R., Aronson, D., Brugieres, L., Brock, P., . . . Plaschkes, J. (2004). Risk-adapted treatment for childhood hepatoblastoma. final report of the second study of the International Society of Paediatric Oncology--SIOPEL 2. Eur J Cancer, 40(3), 411-421.
Pieters, R., Schrappe, M., De, L. P., Hann, I., De, R. G., Felice, M., . . . Valsecchi, M. G. (2007). A treatment protocol for infants younger than 1 year with acute lymphoblastic leukaemia (Interfant-99): an observational study and a multicentre randomised trial. Lancet, 370(9583), 240-250.
Pinto, N. R., Applebaum, M. A., Volchenboum, S. L., Matthay, K. K., London, W. B., Ambros, P. F., . . . Cohn, S. L. (2015). Advances in Risk Classification and Treatment Strategies for Neuroblastoma. J Clin Oncol, 33(27), 3008-3017. https://doi.org/10.1200/jco.2014.59.4648
Principles and practice of pediatric oncology. (2006). (5 utg.). Philadelphia: Lippincott Williams & Wilkins.
Pritchard-Jones, K., & Pritchard, J. (2004). Success of clinical trials in childhood Wilms' tumour around the world. Lancet, 364(9444), 1468-1470.
Pritchard, J., Brown, J., Shafford, E., Perilongo, G., Brock, P., Dicks-Mireaux, C., . . . Plaschkes, J. (2000). Cisplatin, doxorubicin, and delayed surgery for childhood hepatoblastoma: a successful approach--results of the first prospective study of the International Society of Pediatric Oncology. J Clin Oncol, 18(22), 3819-3828.
Prospective Trial for the Diagnosis and Treatment of Intracranial Germ Cell Tumors (SIOPCNSGCTII). (2011-). [pågående studie]. (NCT01424839). Hentet fra http://clinicaltrials.gov/show/NCT01424839
Puget, S., Garnett, M., Wray, A., Grill, J., Habrand, J. L., Bodaert, N., . . . Sainte-Rose, C. (2007). Pediatric craniopharyngiomas: classification and treatment according to the degree of hypothalamic involvement. J Neurosurg, 106(1 Suppl), 3-12.
Pui, C. H. (2006). Central nervous system disease in acute lymphoblastic leukemia: prophylaxis and treatment. Hematology Am Soc Hematol Educ Program, 142-146.
Pui, C. H., & Evans, W. E. (2006). Treatment of acute lymphoblastic leukemia. N Engl J Med, 354(2), 166-178.
Pui, C. H., Schrappe, M., Ribeiro, R. C., & Niemeyer, C. M. (2004). Childhood and adolescent lymphoid and myeloid leukemia. Hematology Am Soc Hematol Educ Program, 118-145.
Radros, J., All-Eriksson, C., Pal, N., Holm, S., Seregard, S., Soderman, M., . . . Holmin, S. (2018). Intra-arterial chemotherapy for retinoblastoma in Sweden - evaluation of treatment efficacy and complications. Acta ophthalmologica, 96(8), e1040-e1041. https://doi.org/10.1111/aos.13796
Ready, P. D. (2010). Leishmaniasis emergence in Europe. Euro Surveill, 15(10), 19505.
Reddy, A. T., & Packer, R. J. (1999). Chemotherapy for low-grade gliomas. Childs Nerv Syst, 15(10), 506-513.
Reilly, A., Kersun, L. S., McDonald, K., Weinberg, A., Jawad, A. F., & Sullivan, K. E. (2010). The efficacy of influenza vaccination in a pediatric oncology population. J Pediatr Hematol Oncol, 32(5), e177-181. https://doi.org/10.1097/MPH.0b013e3181d869f3
Reinfjell, T., Diseth, T., & Vikan, A. (2007). Barn og kreft: Barns tilpasning til og forståelse av alvorlig sykdom. Tidsskrift for Norsk Psykologforening, 44(6), 724-734.
Reiter, A. (2007). Diagnosis and treatment of childhood non-hodgkin lymphoma. Hematology Am Soc Hematol Educ Program, 285-296.
Reni, M., Gatta, G., Mazza, E., & Vecht, C. (2007). Ependymoma. Crit Rev Oncol Hematol, 63(1), 81-89.
Retningslinjer for smertelindring. (2009). Oslo: Den norske legeforening. Hentet fra https://www.legeforeningen.no/om-oss/publikasjoner/retningslinjer/retningslinjer-for-smertelindring-2009/
Richardson, E. A., Ho, B., & Huang, A. (2018). Atypical Teratoid Rhabdoid Tumour : From Tumours to Therapies. Journal of Korean Neurosurgical Society, 61(3), 302-311. https://doi.org/10.3340/jkns.2018.0061
Robinson, G. W., Orr, B. A., & Gajjar, A. (2014). Complete clinical regression of a BRAF V600E-mutant pediatric glioblastoma multiforme after BRAF inhibitor therapy. BMC Cancer, 14, 258. https://doi.org/10.1186/1471-2407-14-258
Rosengren, B., Monge, O. R., & Flage, T. (1989). Proposal of a new pretreatment clinical TNM-classification of retinoblastoma. Acta Oncol, 28(4), 547-548.
Rubin, L. G., Levin, M. J., Ljungman, P., Davies, E. G., Avery, R., Tomblyn, M., . . . Kang, I. (2014). 2013 IDSA clinical practice guideline for vaccination of the immunocompromised host. Clinical infectious diseases : an official publication of the Infectious Diseases Society of America, 58(3), e44-100. https://doi.org/10.1093/cid/cit684
Rutkowski, S. (2006). Current treatment approaches to early childhood medulloblastoma. Expert Rev Neurother, 6(8), 1211-1221.
Rutkowski, S., Bode, U., Deinlein, F., Ottensmeier, H., Warmuth-Metz, M., Soerensen, N., . . . Kuehl, J. (2005). Treatment of early childhood medulloblastoma by postoperative chemotherapy alone. N Engl J Med, 352(10), 978-986.
Rutter's child and adolescent psychiatry. (2008). (5 utg.). Oxford: Blackwell Publ.
Rønning, C., Sundet, K., Due-Tønnessen, B., Lundar, T., & Helseth, E. (2005). Persistent cognitive dysfunction secondary to cerebellar injury in patients treated for posterior fossa tumors in childhood. Pediatr Neurosurg, 41(1), 15-21.
Saghafian-Hedengren, S., Soderstrom, I., Sverremark-Ekstrom, E., & Nilsson, A. (2018). Insights into defective serological memory after acute lymphoblastic leukaemia treatment: The role of the plasma cell survival niche, memory B-cells and gut microbiota in vaccine responses. Blood reviews, 32(1), 71-80. https://doi.org/10.1016/j.blre.2017.08.009
Sandven-Thrane, S. (2012). Leukokori er retinoblastom til det motsatte er bevist. Tidsskr Nor Laegeforen, 132(18), 2042. https://doi.org/10.4045/tidsskr.12.0931
Schellong, G., Dorffel, W., Claviez, A., Korholz, D., Mann, G., Scheel-Walter, H. G., . . . Ruhl, U. (2005). Salvage therapy of progressive and recurrent Hodgkin's disease: results from a multicenter study of the pediatric DAL/GPOH-HD study group. J Clin Oncol, 23(25), 6181-6189.
Schmiegelow, K., Forestier, E., Hellebostad, M., Heyman, M., Kristinsson, J., Soderhall, S., & Taskinen, M. (2010). Long-term results of NOPHO ALL-92 and ALL-2000 studies of childhood acute lymphoblastic leukemia. Leukemia, 24(2), 345-354.
Schrauder, A., Henke-Gendo, C., Seidemann, K., Sasse, M., Cario, G., Moericke, A., . . . Wessel, A. (2007). Varicella vaccination in a child with acute lymphoblastic leukaemia. Lancet, 369(9568), 1232. https://doi.org/10.1016/s0140-6736(07)60567-4
Schrey, D., Carceller Lechon, F., Malietzis, G., Moreno, L., Dufour, C., Chi, S., . . . Zacharoulis, S. (2016). Multimodal therapy in children and adolescents with newly diagnosed atypical teratoid rhabdoid tumor: individual pooled data analysis and review of the literature. J Neurooncol, 126(1), 81-90. https://doi.org/10.1007/s11060-015-1904-0
Schroeder, T. M., Chintagumpala, M., Okcu, M. F., Chiu, J. K., Teh, B. S., Woo, S. Y., & Paulino, A. C. (2008). Intensity-modulated radiation therapy in childhood ependymoma. Int J Radiat Oncol Biol Phys, 71(4), 987-993. https://doi.org/10.1016/j.ijrobp.2007.11.058
Schulz, H., Rehwald, U., Morschhauser, F., Elter, T., Driessen, C., Rudiger, T., . . . Reiser, M. (2008). Rituximab in relapsed lymphocyte-predominant Hodgkin lymphoma: long-term results of a phase 2 trial by the German Hodgkin Lymphoma Study Group (GHSG). Blood, 111(1), 109-111.
Shields, C. L., Mashayekhi, A., Cater, J., Shelil, A., Meadows, A. T., & Shields, J. A. (2004). Chemoreduction for retinoblastoma. Analysis of tumor control and risks for recurrence in 457 tumors. American journal of ophthalmology, 138(3), 329-337. https://doi.org/10.1016/j.ajo.2004.04.032
Shields, C. L., Meadows, A. T., Shields, J. A., Carvalho, C., & Smith, A. F. (2001). Chemoreduction for retinoblastoma may prevent intracranial neuroblastic malignancy (trilateral retinoblastoma). Arch Ophthalmol, 119(9), 1269-1272.
Smedegaard, L. M., Poulsen, A., Kristensen, I. A., Rosthoj, S., Schmiegelow, K., & Nygaard, U. (2016). Varicella Vaccination of Children With Leukemia Without Interruption of Maintenance Therapy: A Danish Experience. Pediatr Infect Dis J, 35(11), e348-e352. https://doi.org/10.1097/inf.0000000000001279
Smeland, S., Bielack, S. S., Whelan, J., Bernstein, M., Hogendoorn, P., Krailo, M. D., . . . Marina, N. (2019). Survival and prognosis with osteosarcoma: outcomes in more than 2000 patients in the EURAMOS-1 (European and American Osteosarcoma Study) cohort. Eur J Cancer, 109, 36-50. https://doi.org/10.1016/j.ejca.2018.11.027
Sposto, R., Ertel, I. J., Jenkin, R. D., Boesel, C. P., Venes, J. L., Ortega, J. A., . . . Hammond, D. (1989). The effectiveness of chemotherapy for treatment of high grade astrocytoma in children: results of a randomized trial. A report from the Childrens Cancer Study Group. J Neurooncol, 7(2), 165-177. https://doi.org/10.1007/bf00165101
Spreafico, F., Pritchard Jones, K., Malogolowkin, M. H., Bergeron, C., Hale, J., de Kraker, J., . . . Graf, N. (2009). Treatment of relapsed Wilms tumors: lessons learned. Expert Rev Anticancer Ther, 9(12), 1807-1815. https://doi.org/10.1586/era.09.159
Stiller, C. (2007). Chilhood Cancer in Britain: incidence, survival, mortality. Oxford: Oxford University Press.
Stokland, T., Liu, J. F., Ironside, J. W., Ellison, D. W., Taylor, R., Robinson, K. J., . . . Walker, D. A. (2010). A multivariate analysis of factors determining tumor progression in childhood low-grade glioma: a population-based cohort study (CCLG CNS9702). Neuro Oncol, 12(12), 1257-1268.
Strauss, R., Neureiter, D., Westenburger, B., Wehler, M., Kirchner, T., & Hahn, E. G. (2004). Multifactorial risk analysis of bone marrow histiocytic hyperplasia with hemophagocytosis in critically ill medical patients--a postmortem clinicopathologic analysis. Crit Care Med, 32(6), 1316-1321.
Sturm, D., Pfister, S. M., & Jones, D. T. W. (2017). Pediatric Gliomas: Current Concepts on Diagnosis, Biology, and Clinical Management. J Clin Oncol, 35(21), 2370-2377. https://doi.org/10.1200/jco.2017.73.0242
Swanson, E. L., Amdur, R. J., Morris, C. G., Galloway, T. J., Marcus, R. B., Jr., Pincus, D. W., & Smith, A. (2011). Intracranial ependymomas treated with radiotherapy: long-term results from a single institution. J Neurooncol, 102(3), 451-457. https://doi.org/10.1007/s11060-010-0344-0
Syse, A. (2008). Does cancer affect marriage rates? Journal of cancer survivorship : research and practice, 2(3), 205-214. https://doi.org/10.1007/s11764-008-0062-1
Timmermann, B., Kortmann, R. D., Kuhl, J., Rutkowski, S., Meisner, C., Pietsch, T., . . . Bamberg, M. (2006). Role of radiotherapy in supratentorial primitive neuroectodermal tumor in young children: results of the German HIT-SKK87 and HIT-SKK92 trials. J Clin Oncol, 24(10), 1554-1560.
Tsang, D. S., Murray, L., Ramaswamy, V., Zapotocky, M., Tabori, U., Bartels, U., . . . Laperriere, N. (2019). Craniospinal irradiation as part of re-irradiation for children with recurrent intracranial ependymoma. Neuro Oncol, 21(4), 547-557. https://doi.org/10.1093/neuonc/noy191
Tsang, D. S., Oliveira, C., Bouffet, E., Hawkins, C., Ramaswamy, V., Yee, R., . . . Laperriere, N. (2019). Repeat irradiation for children with supratentorial high-grade glioma. Pediatr Blood Cancer, 66(9), e27881. https://doi.org/10.1002/pbc.27881
Turaka, K., Shields, C. L., Meadows, A. T., & Leahey, A. (2012). Second malignant neoplasms following chemoreduction with carboplatin, etoposide, and vincristine in 245 patients with intraocular retinoblastoma. Pediatr Blood Cancer, 59(1), 121-125. https://doi.org/10.1002/pbc.23278
Van Dongen-Melman, J. E., Pruyn, J. F., De, G. A., Koot, H. M., Hahlen, K., & Verhulst, F. C. (1995). Late psychosocial consequences for parents of children who survived cancer. J Pediatr Psychol., 20(5), 567-586.
van Tilburg, C. M., van Gent, R., Bierings, M. B., Otto, S. A., Sanders, E. A., Nibbelke, E. E., . . . Tesselaar, K. (2011). Immune reconstitution in children following chemotherapy for haematological malignancies: a long-term follow-up. Br J Haematol, 152(2), 201-210. https://doi.org/10.1111/j.1365-2141.2010.08478.x
Vanan, M. I., & Eisenstat, D. D. (2015). DIPG in Children - What Can We Learn from the Past? Frontiers in oncology, 5, 237. https://doi.org/10.3389/fonc.2015.00237
Wallace, W. H., Blacklay, A., Eiser, C., Davies, H., Hawkins, M., Levitt, G. A., & Jenney, M. E. (2001). Developing strategies for long term follow up of survivors of childhood cancer. BMJ, 323(7307), 271-274.
Wiegering, V., Frank, J., Freudenberg, S., Morbach, H., Schlegel, P. G., Eyrich, M., & Winkler, B. (2014). Impaired B-cell reconstitution in children after chemotherapy for standard or medium risk acute precursor B-lymphoblastic leukemia. Leukemia & lymphoma, 55(4), 870-875. https://doi.org/10.3109/10428194.2013.816423
Wong, J. R., Morton, L. M., Tucker, M. A., Abramson, D. H., Seddon, J. M., Sampson, J. N., & Kleinerman, R. A. (2014). Risk of subsequent malignant neoplasms in long-term hereditary retinoblastoma survivors after chemotherapy and radiotherapy. J Clin Oncol, 32(29), 3284-3290. https://doi.org/10.1200/JCO.2013.54.7844
Wright, K. D., Green, D. M., & Daw, N. C. (2009). Late effects of treatment for wilms tumor. Pediatr Hematol Oncol, 26(6), 407-413.
Yousef, Y. A., Al-Hussaini, M., Mehyar, M., Sultan, I., Jaradat, I., AlRawashdeh, K., . . . Nawaiseh, I. (2015). PREDICTIVE VALUE OF TNM CLASSIFICATION, INTERNATIONAL CLASSIFICATION, AND REESE-ELLSWORTH STAGING OF RETINOBLASTOMA FOR THE LIKELIHOOD OF HIGH-RISK PATHOLOGIC FEATURES. Retina (Philadelphia, Pa.), 35(9), 1883-1889. https://doi.org/10.1097/iae.0000000000000547
Yu, A. L., Gilman, A. L., Ozkaynak, M. F., London, W. B., Kreissman, S. G., Chen, H. X., . . . Sondel, P. M. (2010). Anti-GD2 antibody with GM-CSF, interleukin-2, and isotretinoin for neuroblastoma. N Engl J Med, 363(14), 1324-1334. https://doi.org/10.1056/NEJMoa0911123
Zacharoulis, S., Ashley, S., Moreno, L., Gentet, J. C., Massimino, M., & Frappaz, D. (2010). Treatment and outcome of children with relapsed ependymoma: a multi-institutional retrospective analysis. Childs Nerv Syst, 26(7), 905-911. https://doi.org/10.1007/s00381-009-1067-4
Zacharoulis, S., & Moreno, L. (2009). Ependymoma: an update. J Child Neurol, 24(11), 1431-1438.
Zeller, B., Gustafsson, G., Forestier, E., Abrahamsson, J., Clausen, N., Heldrup, J., . . . Hasle, H. (2005). Acute leukaemia in children with Down syndrome: a population-based Nordic study. Br J Haematol, 128(6), 797-804.
Zhang, J., Walsh, M. F., Wu, G., Edmonson, M. N., Gruber, T. A., Easton, J., . . . Downing, J. R. (2015). Germline Mutations in Predisposition Genes in Pediatric Cancer. N Engl J Med, 373(24), 2336-2346. https://doi.org/10.1056/NEJMoa1508054
Zsiros, J., Brugieres, L., Brock, P., Roebuck, D., Maibach, R., Zimmermann, A., . . . Perilongo, G. (2013). Dose-dense cisplatin-based chemotherapy and surgery for children with high-risk hepatoblastoma (SIOPEL-4): a prospective, single-arm, feasibility study. The Lancet. Oncology, 14(9), 834-842. https://doi.org/10.1016/s1470-2045(13)70272-9
Aarsen, F. K., Van Dongen, H. R., Paquier, P. F., Van, M. M., & Catsman-Berrevoets, C. E. (2004). Long-term sequelae in children after cerebellar astrocytoma surgery. Neurology, 62(8), 1311-1316.