









Generell veileder i pediatri
8. Hjerte- og karsykdommer
8.30 Marfan syndrom og arvelige aortopatier
Sist faglig oppdatert: 17.06.2025
Emil Stefors, Mads N. Holten-Andersen, Christian Neukamm, Kirsten Krogh-Sørensen, Benedicte Paus og Henrik Holmstrøm
Bakgrunn
Arvelige aortopatier er en gruppe genetiske bindevevssykdommer som gir risiko for aortadilatasjon, disseksjon og ruptur. Samlebetegnelsen er Heritable Thoracic Aortic Diseases (HTAD) og flere har blitt genetisk karakterisert de siste årene. Det finnes ingen etablerte guidelines for oppfølging av aortopatier og dette kapitlet er i stor grad basert på et nylig publisert Scientific Statement fra AHA, tilpasset norske forhold.
Risikoen for komplikasjoner er avhengig av den genetiske årsaken, men det er også store fenotypiske variasjoner. Familiehistorien er derfor viktig i vurderingen av risiko. Bikuspid aortaklaff (BAV) er en aortopati med svakere genetisk komponent og blir omtalt i et eget kapittel. Risikoen for aortakomplikasjoner ved HTAD øker hos personer som samtidig har BAV.
Marfan syndrom er en autosomal dominant arvelig bindevevssykdom med prevalens på 5–7/100 000 og årlig insidens på 1/100 000. Flertallet (> 90 %) har påvisbar sykdomsgivende variant av fibrillin-genet (FBN1). HTAD omfatter mange andre geno- og fenotyper, og de mest relevante genene (tilstandene) er:
- FBN1 (Marfan syndrom)
- TGFB1 og -2, TGFBR1 og -2 og SMAD2 og -3 (Loeys-Dietz syndrom type 1–6)
- COL3A1 (Vaskulært Ehlers-Danlos syndrom)
- ACTA2, MYH11, MYLK, LOX, FOX, EFEMP2 og PRKG1 (ikke-syndromale HTAD)
Selv om risikoen for aortakomplikasjoner varierer mellom tilstandene, er diagnostikk, oppfølging og behandling såpass like at de her blir omtalt samlet. Marfan syndrom blir beskrevet mer detaljert, fordi syndromet omfatter flere andre organsystemer og fordi det er hyppigst og best kjent.
Symptomer og funn
Typiske, kliniske kjennetegn og andre organmanifestasjoner enn aortopati forekommer ofte ved Marfan syndrom, men sees også ved enkelte av de andre. Det er stor variasjonsbredde med alt fra isolerte Marfan-trekk til (ytterst sjelden) neonatal presentasjon med alvorlig, rask progressiv sykdom med multiorgan-affeksjon. Hovedårsaken til morbiditet og mortalitet er dilatasjon og disseksjon av aorta, noe som er uhyre sjeldent før voksen alder.
Følgende karakteristika kan forekomme ved Marfan syndrom:
- Hjerte/kar: Aortarotdilatasjon, aortainsuffisiens, dilatasjon av andre segmenter av aorta (eller andre arterier i toraks/hals/bekkenregion), mitralprolaps/-insuffisiens
- Øye: Synsproblemer (spesielt linseluksasjon, myopi, netthinneløsning, katarakt, glaukom)
- Toraks: Brystkasse-deformitet (fugle-, traktbryst), spontan pneumotoraks
- Nervesystem: Utvidelse av duralsekken (dural ektasi)
- Skjelett: Leptosom kroppsbygning (lang og tynn, relativt smal brystkasse, lange ben og lange, smale hender og føtter), øket lengdevekst, lange armer/ben/fingre, skoliose, kyfose, hypermobile ledd, plattfot, protrusio acetabuli
- Hud/hinner: Striae, residiverende hernier
Diagnostikk og utredning
Spørsmål om HTAD oppstår vanligvis i en av følgende situasjoner:
- Genetisk familieutredning (vanligst hos barn)
- Utredning i forbindelse med en akutt kardiovaskulær hendelse (oftest voksne pasienter, unntaksvis ungdommer)
- Klinisk mistanke om Marfan syndrom (eller ytterst sjelden, en av de andre tilstandene)
- Tilfeldig påvist aortadilatasjon ved kardiologisk undersøkelse
Ca. 25 % av Marfan-tilfellene er oppgitt å være nyoppståtte, dvs. uten sykdom hos foreldrene. Diagnostikken ved Marfan syndrom er basert på de reviderte Ghent kriteriene (2010). Diagnosen kan stilles ved kombinasjoner av aortarotdilatasjon, linseektopi, systemiske funn, skjelettfunn og sykdomsgivende mutasjon i FBN1, avhengig av familieanamnese.
Ingen familieanamnese for Marfan:
1. Aortarot-dilatasjon (Z-skår ≥ 2, eller aortarot-disseksjon) OG linseektopi*
2. Aortarot-dilatasjon (Z-skår ≥ 2, eller aortarot-disseksjon) OG FBN1 sykdomsgivende variant
3. Aortarot-dilatasjon (Z-skår ≥ 2, eller aortarot-disseksjon) OG system-skår > 7*
4. Linseektopi OG FBN1 sykdomsgivende variant assosiert med aortarotdilatasjon/-disseksjon
Positiv familieanamnese (1. gradsslektning som oppfyller en av 1–4) OG en av følgende:
5. Linseektopi
6. System-skår ≥ 7*
7. Aortarot-dilatasjon (Z-skår > 3 ved alder < 20 år, eller aortarot-disseksjon)*
*Utelukkelse av andre beslektede bindevevssyndromer samt genetiske forandringer. For beregning av system-skår samt Z-skår for aortaroten, se: www.Marfan.org/dx.
Genetisk utredning
Genetisk utredning er avgjørende for HTAD-diagnostikken og det finnes et panel for genetiske bindevevssykdommer. Man må skille på diagnostisk og prediktiv genetisk testing, og ved prediktiv testing må familiene henvises til genetisk veiledning før prøven blir tatt. Hos barn er det naturlig at diagnostikken foregår på barneavdelingene, men i noen tilfeller tar fastlegene dette ansvaret. Det viktigste er at oppgavefordelingen blir avklart.
Barn med påvist mutasjon følges opp og behandles i tråd med retningslinjene nedenfor.
I klinisk praksis vil mange barn ikke oppfylle de reviderte Ghent-kriteriene, men vokse seg inn i diagnosen med tiden. Derfor er bruk av godt klinisk skjønn viktig. Barna skal følges som om de har Marfan syndrom inntil man er sikker på å kunne utelukke diagnosen.
Noen ganger påvises en VUS (genetisk variant med usikker betydning) hos barna, eller det er påvist en slik variant hos en av foreldrene. Disse barna bør følges klinisk til man er sikker på om mutasjonen er sykdomsgivende eller ikke. Undersøkelse av familiemedlemmer kan i noen tilfeller bidra til å oppklare betydningen av genforandringen.
Potensielle genbærere som ikke er genetisk utredet (f.eks. dersom foresatte ikke ønsker genundersøkelse av barn under 16 år) bør som minimum følges hvert 2. år med ekkokardiografi og øyelegeundersøkelse.
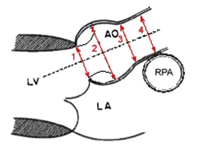

Ekkokardiografi
Ekko-apparatene har integrerte referansematerialer som er forskjellige og ikke sammenlignbare. Det er derfor avgjørende at aortarotens Z-skår angis etter samme kriterier, og vi har valgt å bruke den amerikanske Marfanforeningens kalkulator som er basert på indre diameter i systole (https://www.marfan.org/dx/zscore). Man må unngå koronaravgangene, og finne et symmetrisk bilde med god definisjon av veggkonturene. Merk at Z-skårene kan være misvisende hos barn med avvikende vekt, særlig utenom persentilkurvene. Førstegangs-undersøkelse skal omfatte en full barnekardiologisk protokoll med særlig fokus på aortaroten og mitralklaffen (prolaps, insuffisiens). I videre oppfølgning kan undersøkelsen begrenses til relevante avsnitt (aortadimensjoner, mitral- og aortaklaff, ventrikkel-dimensjoner og funksjon).
MR-undersøkelser
Hovedindikasjon for MR er kartlegging av aorta hos alle med påvist dilatasjon av aortaroten på ekko eller alvorlig familiehistorie. MR foretrekkes ved diagnostikk hos barn for å redusere stråledosene og generelt bør den utsettes til den kan gjennomføres uten narkose. Første MR gjennomføres ved 13–14 års alder eller tidligere på spesiell indikasjon (f.eks. økende aorta- eller mitrallekkasje, uttalt/raskt progredierende dilatasjon eller mistanke om (distal) aortadilatasjon/disseksjon som ikke avklares ved ekko). Behov for videre MR-kontroll vurderes ut fra individuelle funn. MR i narkose gjennomføres på spesiell indikasjon. I utvalgte tilfeller kan noen ganger CT være et aktuelt alternativ, særlig ved preoperativ utredning og/eller mistenkt disseksjon. MR kan i noen tilfeller også være indisert ved diagnostikk av andre problemstillinger som protrusio acetabuli og dural ektasi.
Det er viktig at MR-undersøkelser gjøres etter spesielle protokoller og vurderes av et fagmiljø med erfaring. (Barneradiologisk seksjon ved OUS kan gi råd og anvisninger).
Behandling og oppfølging
| Behandlingsprotokoll | Grad av aorta-dilatasjon | |||
| Ingen | Mild | Moderat | Alvorlig | |
| Alder < 16 år (Z-score) | < 2 | ≥ 2 til < 3,5 | ≥ 3,5 til < 5 | ≥ 5 |
| Alder > 16 år (cm) | < 3,5 | ≥ 3,5 til < 4 | ≥ 4 til < 4,5 | ≥ 4,5 |
| Omtrentlig kontrollintervall (md.) | 24 | 12–18 | 12 | 6 |
| Behandlingstrinn | Ingen behandling | Ett medikament | Dobbeltbehandling bør vurderes | To medikamenter |
Faktorer som trekker i retning av hyppigere kontroller og mer behandling:
- Påvist dilatasjon av aortaroten ved antatt aggressive tilstander (mutasjonene FBN1 og TGFBR1 og -2, alvorlig familiehistorie; flere personer eller yngre personer med kardiovaskulære komplikasjoner som aortaaneurisme, -disseksjon og -kirurgi)
- Uttalte organmanifestasjoner utenom hjertet
- Rask vekst av aortaroten (≥ 0,8 cm/år før 2 års alder eller > 0,5 cm/år etter det)
- Bikuspid aortaklaff
- Aorta- eller mitralklaffelekkasje
Medikamentell behandling
I tråd med internasjonale guidelines anbefaler vi i Norge medikamentell behandling når aortadilatasjon blir påvist (se tabellen over). Man kan forvente å oppnå ca. 10 års utsettelse av behovet for kirurgi, og dette skal veies mot eventuelle ulemper med medisineringen. Dette må forklares og diskuteres med familien.
Betablokkere og angiotensin-II reseptorantagonister (A2-blokkere) har likeverdig effekt, og det er ingen sikker forskjell mellom forskjellige preparater. Av A2-blokkerne har Irbesartan har vært mest brukt de siste årene, men Losartan er et likeverdig alternativ og finnes som mikstur for barn. Doseringen er for Losartan ca. 1 mg/kg/d (max 50 mg ved opp til 50 kg vekt og max 100 mg ved over 50 kg vekt. Halv dose første 2–4 uker). Se Koble for ytterligere info.
Av betablokkere er Metoprolol depottabletter mest brukt i Norge, og vanlig dosering er ca. 1 mg/kg/døgn (halv dose første 2–4 uker). Målsettingen er ca. 20 % reduksjon i makspuls sammenlignet med før behandlingsoppstart. Hos større barn kan dette prøves ut ved tredemølletest før og etter oppstart, men dette er en ressurskrevende undersøkelse som ikke kan tilbys generelt i denne situasjonen. Man kan også få en oppfattelse om pulsnivået ved 24-timers EKG-registrering. Dette forutsetter at registreringene gjennomføres på dager med likt aktivitetsnivå og at barnet virkelig oppnår maksimalt belastende aktivitet. Hos barn under 4–5 år er dette sjelden tilfelle, slik at undersøkelsen kan bli villedende. Pulsklokkenes rolle i slik monitorering er foreløpig uavklart.
Kombinasjon av betablokker og A2-blokkere brukes som angitt i tabellen ovenfor.
Fluorokinoloner bør ikke brukes ved HTAD pga. risiko for forverring av aortopatien. ADHD behandling kan gis til barn ut fra vanlige prinsipper.
Aktivitetstilpasning
Barn med HTAD bør delta regelmessig i fysisk aktivitet som alle andre barn. Frem til 13–14 års alder gis begrensninger bare i helt spesielle tilfeller. Risikoen for linseluksasjon og netthinneløsning tilsier at man bør fraråde fysisk kontaktsport samt anbefale bruk av beskyttende briller ved aktivitet med liten ball. Hos ungdommer med spesielt høy kardiovaskulær risiko frarådes aktivitet ved høy/maksimal intensitet (anaerobt nivå), isometrisk trening og styrketrening med høy belastning. Deltakelse i idrett på konkurransenivå fra tenåringsalder bør drøftes med barnekardiolog.
Kirurgisk behandling
Vurderes individuelt, blant annet basert på familiehistorie, påvist mutasjon og klinisk utvikling. Etablerte kriterier ved Marfan syndrom er aortarot diameter ≥ 45–50 mm, hurtig økende dilatasjon, økende aortainsuffisiens eller behandlingskrevende mitralinsuffisiens. Hos barn og ungdommer er det ofte klaffelekkasje som utløser kirurgi. For at man skal unngå implantasjon av mekanisk aortaventil, kan operasjonen gjennomføres selv om diametermålet ikke er oppnådd. Dersom det vurderes å være høy risiko for akutt disseksjon, kan kirurgi vurderes på lavere diameter, helt ned mot 40 mm, særlig ved raskt økende dilatasjon. Guidelines lister en lang rekke risikofaktorer som bør vurderes ved de ulike genmutasjoner, og beslutninger bør tas i multidisiplinære team og i samråd med pasient og pårørende.
Oppfølgning
Individuelt tilpasset tverrfaglig oppfølgning. Genetiker ifm. diagnose. Første kontroll 6 måneder etter diagnose for å vurdere stabilitet av aortadilatasjon. Heretter årlige kontroller med ekko-cor, puls/BT, EKG, øyelege. Etter behov: ortoped, fysioterapeut, MR/CT, 24-timers EKG (for vurdering av medikamentell behandlingseffekt eller ved mistanke om arytmi). For oppfølgning og tiltak ved mitral-prolaps/insuffisiens, øyeproblemer, ortopediske problemstillinger: se også www.marfan.org/dx. Det anbefales at barn med Marfan syndrom henvises til vurdering ved universitetssykehus enten for diagnostisering eller i løpet av det første året etter etablert diagnose. Videre oppfølgning kan foregå på lokalsykehusene og behov for henvisning til universitetssykehus vurderes individuelt. Sentral kontroll bør uansett gjennomføres ved indikasjon for endret medisinering og i forbindelse med MR i 12–14 års-alderen. Oppfølging av klaffeproblematikk vurderes på selvstendig grunnlag.
Det er opprettet en tverrfaglig spesialistklinikk («Bindevevspoliklinikken» ved Thoraxkirurgisk avdeling, OUS) for behandling og oppfølging av personer med HTAD. Den er medlem i det Europeiske referansenettverket for HTAD (www.vascern.eu), og fagmiljøet har omfattende klinisk erfaring med arvelige bindevevssykdommer.
Pasienter med kjent eller velbegrunnet mistenkt HTAD kan henvises til Bindevevspoliklinikken. Det er ønskelig at pasienter henvises ved overgang fra barn- til voksen-spesialisthelsetjeneste for å få bred informasjon om alle sider ved å leve med en arvelig tilstand.
TRS Sunnaas som er en del av Bindevevsklinikken, er et nasjonalt kompetansesenter med ansvar for Marfan syndrom og andre genetiske aortopatier. Senteret er tverrfaglig og kan blant annet gi råd om medisinske og psykologiske forhold, fysisk aktivitet, dagligliv, skole, arbeidsliv og rettigheter. TRS har ikke oppfølgningsansvar for pasienter med Marfan syndrom, men kan kontaktes av brukerne, deres pårørende eller fagpersoner ved behov. Informasjon om diagnosen finnes på TRS sine nettsider.
Referanser
- Morris SA, et al. Cardiovascular Management of Aortopathy in Children: A Scientific Statement from the American Heart Association, Circ 2024; 149:e00–e00
- Milewicz DM, et al. Marfan Syndrome, Nature Rev 2021; 7:1-24
- Loeys BL, et al. The revised Ghent nosology for the Marfan syndrome. J Med Genet 2010; 47:476-85
- Pitcher A, et al. ARBs and β-blockers in Marfan syndrome: an individual patient data meta-analysis of randomised trials. Lancet 2022; 400:822-31
Tidligere versjoner
2018: Mads N. Holten-Andersen, Henrik Holmstrøm, Christian Neukamm og Nina Riise