









Nyfødtveileder
10 Glukose, elektrolytter og metabolsk sykdom
10.8 Hyperammonemi og ureasyklusdefekter
Sist faglig oppdatert: 27.11.2024
Andreas Øberg, Claus Klingenberg og Trine Tangeraas
Bakgrunn
- Ammoniakk oppstår bl.a. etter nedbryting av proteiner og produseres av tarmbakterier.
- Omtrent 90 % av ammoniakk som produseres vil gå inn i levercellene og der omdannes til urea ved hjelp av ulike enzymer i ureasyklus (se figur under).
- Vanligste årsaker til hyperammonemi i nyfødtperioden er angitt i tabellen under.
- Hyperammonemi er aldri til stede ved fødsel da ammoniakk går over placenta og detoksifiseres i mors lever. De første symptomene på hyperammonemi/ureasyklusdefekt oppstår fra 2. levedøgn og er initialt ofte vage i form av redusert sugeevne, søvnighet, slapphet og ev. hyperventilering (respiratorisk alkalose).
- Ubehandlet hyperammonemi > 200–250 μmol/L er assosiert med alvorlig CNS-skade og mortalitet.
Tabell: Årsaker til hyperammonemi i nyfødtperioden
| Årsak | Mekanisme |
| Alvorlig sykt barn | Sannsynligvis multifaktorielt betinget Sjelden ammoniakk > 150 μmol/L |
| Dekompensert leversvikt | Redusert antall funksjonelle hepatocytter gir redusert ureasyklus Sjelden ammoniakk > 300 μmol/L |
| Ureasyklusdefekter | Ofte «frisk» lever, men defekt i ett av enzymene i ureasyklus Ammoniakk > 200 μmol/L og opp til 2000 μmol/L |
| Organisk acidurier | Opphopning av organiske syrer hemmer enzymene NAGS og CPS1 i ureasyklus Ammoniakk > 200 μmol/L og opp til 2000 μmol/L |
| Medikamentindusert hyperammonemi | Overproduksjon av ammoniakk. Sjeldent i nyfødtperioden. Typiske medikamenter: Valproatsyre, karbamazepin, salicylater, PEG-asperginase. |
- Barn med hyperammonemi > 150–200 μmol/L uten metabolsk acidose eller alvorlig leversvikt (+ ofte initialt med en respiratorisk alkalose) har trolig en urasyklusdefekt (også kalt primær hyperammonemi). Obs. nyfødte barn med ureasyklusdefekter kan utvikle metabolsk acidose sent i forløpet!
- Barn med hyperammonemi > 150–200 μmol/L med metabolsk acidose (anion gap > 20 og/eller hypoglykemi og/eller ketoner i blod eller urin) gir mistanke organisk aciduri (f.eks. propionsyreemi, metylmalonsyreemi etc.). Fettsyreoksidasjonsdefekter og mitokondriell sykdom kan også ha forhøyet ammoniakk, men sjeldent verdier > 300 μmol/L og de har som regel ingen ketonuri.
Det blir i 2025 startet nyfødtscreening for 3 av 6 ureasyklusdefekter (ASS, ASL og ARG1), men rundt 40 % av barn med ureasyklusdefekt vil debutere med symptomer før svar på nyfødtscreening foreligger, og de proksimale defektene (NAGS, OTC, CPS1) er ikke inkludert i nyfødtscreeningen.
Tidlig behandling er avgjørende for å hindre/minimalisere en irreversibel hjerneskade.
Første ammoniakkrise (> 200–250 μmol/L) kan ta lang tid å reversere og vil ofte kunne eskalere tross optimalisert konvensjonell behandling. Alle nyfødte med alvorlig hyperammonemi bør derfor tidlig diskuteres med nyfødtbakvakt/metabolsk lege på OUS-Rikshospitalet mtp. indikasjon for overflytting til hemodiafiltrasjon.
Figur: Ureasyklus med enzymer angitt i blå skrift
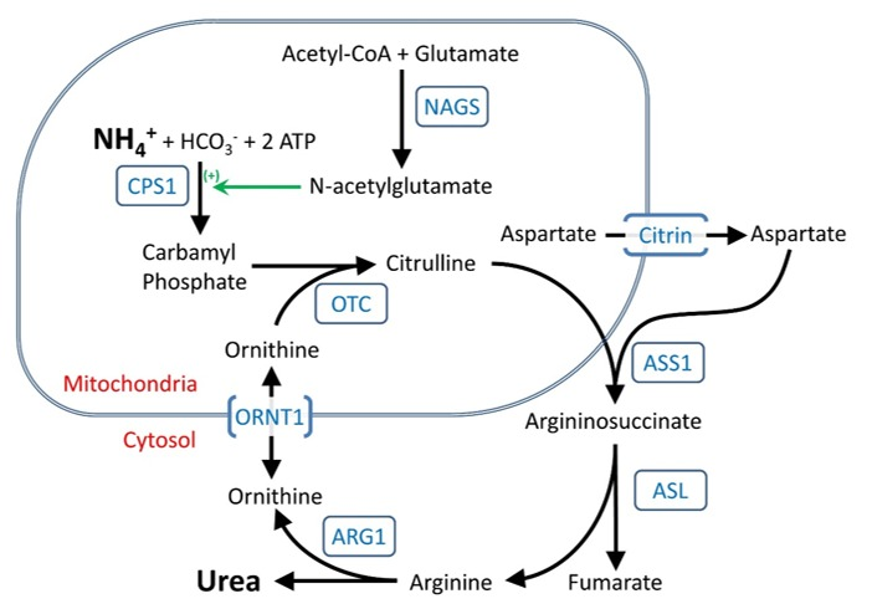
Argininosuksinatsyntetase (ASS), Argininsuksinatlyase (ASL), Arginase-1 defekt (ARG1)
Symptomer og klinikk
- Pasienter med ureasyklusdefekter er oftest friske ved fødsel før de fra annet levedøgn og utover kan utvikle vage/uspesifikke symptomer som slapphet, spisevansker, problemer med termoregulering og hyperventilering (respiratorisk alkalose!).
- Ofte vekttap > 10 %.
- Kramper er vanlig, men et sent symptom.
- Deretter vil hjerneødem og forhøyet ammoniakk føre til progredierende encefalopati/koma og ev. død.
- Defekter tidlig i ureasyklus (NAGS, CPS, OTC) gir typisk de høyeste ammoniakkverdiene.
Hos omtrent halvparten av pasientene med ureasyklusdefekter oppstår symptomene etter nyfødtperioden og er da generelt mindre alvorlige. Disse pasientene kan oppleve perioder sløvhet, slapphet, oppkast, magesmerter, adferdsvansker og kognitiv dysfunksjon. Arginasedefekt debuterer snikende over år og leder vanligvis til varierende grad av kognitiv dysfunksjon og spastisk diplegi uten uttalt hyperammonemi.
Diagnostikk og utredning
Bestill alltid ammoniakk i plasma som ø-hjelp hos barn med encefalopati av uklar årsak.
Alltid obs. hos nyfødte med uklar årsak til respiratorisk alkalose som kan være et tidlig tegn på hyperammonemi!
NB. Hos friske nyfødte kan ammoniakkverdier opp til 110 μmol/L være normalt, men verdier > 150 μmol/L er suspekt på metabolsk sykdom. Obs. falsk forhøyede ammoniakkverdier ved feil prøvetaking (skvising fra vevet, kapillær prøve etc.). Prøven må derfor tas venøst eller fra arteriekran, settes rett på is og analyseres raskt!
Øvrig utredning
- Hematologiske prøver, CRP, SBS, laktat, anion gap, INR, leverprøver
- Sendeprøver OUS for metabolske analyser: 0,5 ml heparinplasma og 0,5 ml EDTA plasma samt 5–10 ml urin til metabolsk screening, fryses raskt.
- Ultralyd caput - ødem?
- EDTA blod til aktuell genetisk avdeling, men ikke så hast.
Ved mistanke om ureasyklusdefekt kan nyfødtscreeningen kontaktes for hurtig-genetikk på barnets filterkort (forskningsprosjekt Nyfødtscreeningen). Ring nyfødtscreeningen 23 07 59 61/73 og vakthavende lege ved metabolsk lab: 90927517.
Behandling og oppfølging
- Akuttbehandling består i å stoppe proteintilførsel (maks. 24–36 timer) samtidig som man må sikre adekvat energitilførsel fra glukose og fett for å hindre katabolisme.
- Start glukose 150 mg/ml i.v., initialt 4,5 ml/kg/time (10 mg/kg/min).
- Obs. ta høyde for ganske store mengder i.v. glukose og bruk Glukose 100 mg/ml som utblandingsvæske for «scavenger treatment» med Na-benzoat, Na-fenylbutyrat og argininhydroklorid, spesielt første 2 timer (se tabell neste side).
- Ved ukjent årsak til hyperammonemi bør også kargluminsyre forsøkes. Kargluminsyre virker kun på den ultrasjeldne NAGS defekt, men har ofte også svært god effekt på hyperammonemi sekundært til organiske acidurier.
- Etabler NVK eller sentralvenøs tilgang. Øvrig intensivbehandling følger samme prinsipper som hos andre kritisk syke nyfødte, men obs. høy risiko for hjerneødemutvikling hos pasientgruppen.
Tilstreb 100 kcal/kg/døgn i form av glukose med elektrolytter og Clinoleic/SMOF-lipid 2–3 g/kg/d. Ofte vil glukose-insulindrypp være nødvendig, se neonatal hyperglykemi. Blodsukker ønskes 6–10 mmol/L. Reintroduser protein proteintilskudd etter 24 timer da tap av essensielle aminosyrer vil føre til økt katabolisme og nitrogenfrigjøring.
Hemodiafiltrasjon
Vurderes hos alle nyfødte med ureasyklusdefekt og etableres så raskt som mulig ved alvorlig hyperammonemi (> 250 μmol/L og nevrologisk påvirket barn (encefalopati) parallelt med innsatt behandling (dvs. innen 6 timer) eller ved raskt stigende verdier). Peritoneal dialyse er lite effektivt og brukes kun om hemofiltrasjon ikke er mulig. Utskiftningstransfusjon er ikke effektivt og kan gi økt proteinbelastning.
Farmakologisk behandling (se tabell)
Denne skal aktivere alternative veier for nitrogeneliminasjon («scavenger treametment» med Na-benzoat og/eller Na-fenylbutyrat) samt erstatte manglende arginin pga. stopp i ureasyklus (tilskudd av argininhydroklorid) eller stimulere enzymaktivitet (kargluminsyre).
Ofte er dette ikke tilstrekkelig for å fjerne ammoniakk raskt nok hos alvorlig syke nyfødte med tidlig debut, men det bør startes i påvente av overflytting og ev. hemodiafiltrasjon.
Akuttbehandling
Start med Natriumbenzoat, Argininhydroklorid og Kargluminsyre.
NB! Kargluminsyre er primært utviklet for NAGS-defekter proksimalt i ureasyklus og virker ikke ved distale ureasyklusdefekter. Metningsdose prøves allikevel alltid ved alvorlig hyperammonemi før sikker diagnose er etablert da kargluminsyre også kan ha svært god effekt på alvorlig hyperammonemi sekundært til organiske acidurier.
Tabell: Oversikt over akuttmedikasjon ved alvorlig hyperammonemi
| Medikament | Styrke | Dose/administrasjon | Virkningsmekanisme |
Na-benzoat | Fortynnes med glukose 100 mg/ml til en konsentrasjon Se blandekort | Metningsdose: Vedlikehold: | Ammoniakk bindes til glycin og skilles ut i urin |
Na-fenylbutyrat* (Ambutyrate® 2 g/10 ml = 200 mg/ml) | Fortynnes med glukose 100 mg/ml til en konsentrasjon Se blandekort | Metningsdose: Vedlikehold: | Ammoniakk bindes til glutamin og skilles ut i urin |
| Ammonul®** Kombinasjonspreparat: Na-benzoat/Na-fenylacetat 100 mg/ml | Fortynnes med glukose 100 mg/ml til en konsentrasjon Se blandekort | Metningsdose: Vedlikehold: | |
Argininklorid NAF® NB. Argininklorid NAF inneholder altså Argininhydroklorid | Fortynnes med glukose 100 mg/ml til en konsentrasjon Se blandekort | Metningsdose: Vedlikehold:
| Øker amoniakkutskillelse i ureasyklus og tilfører arginin Surt. Følg pH, ev. gi buffer. Kan gi hudnekrose ved ekstravasering! |
| Kargluminsyre (Ucedane® eller Carbaglu®) Tbl. a 200 mg | 200 mg tabletter Løses i vann, gis p.o. | Metningsdose: Vedlikehold: | Stimulerer N-acetylglutamat syntase (NAGS) i ureasyklus |
*Ikke rutinemessig tilgengelig. **Kun i beredskap ved nyfødt intensiv OUS Rikshospitalet.
Vedlikeholdsbehandling
Diskuteres med OUS-RH, men veiledende kan man fortsette med i.v. dosering fra tabellen inntil ammoniakk-verdiene er normalisert (< 100 μmol/L).
Man kan da gå over til peroral behandling med Na-benzoat og/eller Na-fenylbutyrat/glycerolfenylbutyrat for å fjerne ammoniakk.
- Na-benzoat: < 20 kg opptil 250 mg/kg/d delt på 3 doser. Vekt > 20 kg max 5 g/m2/døgn.
- Glycerylfenylbutyrat mikstur (Ravicti®) mikstur 1,1 g/ml: 9,4 g/m2/døgn, fordelt på 3 doser
- Avhengig av type ureasyklusdefekt gis ev. også:
- Argininhydroklorid (fortynnet til 0,5 mmol/ml): 0,5 mmol/kg x 4, ev. overgang til Arginin sachets (doseposer) eller citrullin.
I tillegg: Sterk proteinredusert diett, ev. med tilskudd av essensielle aminosyrer
Prognose
- Ved hyperammonemi > 2000 μmol/L og/eller klinisk encefalopati > 24 timer er mortalitet og morbiditet (CNS sekveler) generelt sett svært høy, men dette er ofte vanskelig å prognostisere i akuttsituasjon da mye av akutt skade er reversibelt.
- Hvis en katastrofal hyperammonemi unngås er levertransplantasjon (ved 3–12 md. alder) potensielt kurativt ved de fleste ureasyklusdefekter og per nå foretrukket langtids behandlingsstrategi ved de fleste neonatale - og mange av de senere debuterende ureasyklusdefekter.
Referanser
- GeneReviews: Urea Cycle Disorders Overview. Fin oversikt over videre diagnostikk
- Häberle J et al. Suggested guidelines for the diagnosis and management of urea cycle disorders: First revision. J Inherit Metab Dis. 2019; 42: 1192-1230. Fullstendig oversikt om diagnostikk og behandling.
- Burgard P, et al. Neonatal mortality and outcome at the end of the first year of life in early onset urea cycle disorders--review and meta-analysis of observational studies published over more than 35 years. J Inherit Metab Dis 2016; 39: 219-29.
- Häberle J. Primary Hyperammonaemia: Current Diagnostic and Therapeutic Strategies. J Mother Child 2020; 24: 32-38.
- Savy N, et al. Acute pediatric hyperammonemia: current diagnosis and management strategies. Hepat Med 2018; 10: 105-15.
- Lindemann R, et al. Et nyfødt barn med hyperventilasjon. Tidsskr Nor Legeforen 2008; 128: 1535-6.
Tidligere versjoner
2021: Claus Klingenberg, Nils Thomas Songstad, Trine Tangeraas